Multi-objective optimization and quantum hybridization of equivariant deep learning interatomic potentials on organic and inorganic compounds
1 A research team from Terra Quantum AG and collaborators has developed enhanced variants of the Allegro machine learning interatomic potential (MLIP) model, achieving superior accuracy through multi-objective hyperparameter optimization and novel quantum-classical hybrid architectures.
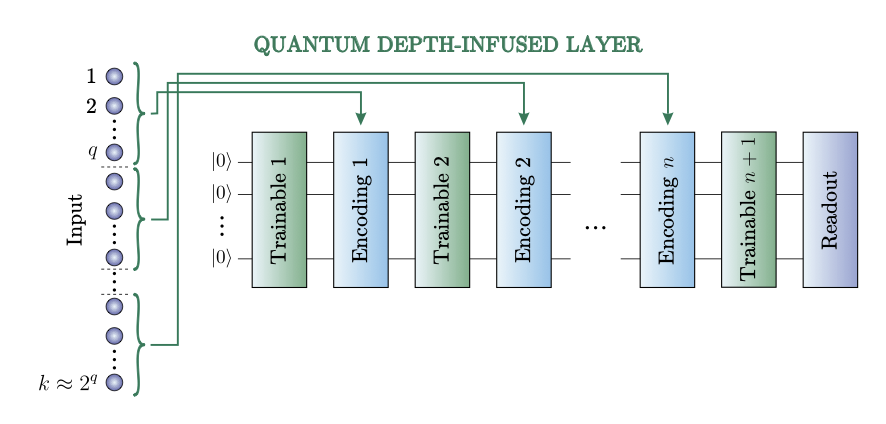
2 The study introduces two key innovations: an extended Allegro model with additional classical MLP layers (Allegro MLP) and a quantum depth-infused variant (Allegro QDI) that incorporates variational quantum circuits into the neural network architecture.
3 Using their implementation of the SAMO-COBRA multi-objective optimization algorithm, the researchers simultaneously optimized for prediction accuracy (force/energy MAE) and inference speed across four diverse datasets: QM9 organic molecules, rMD17 aspirin and benzene subsets, and a newly generated copper-lithium inorganic dataset.
4 The Allegro QDI model demonstrated remarkable performance on the Cu-Li dataset, achieving a 13% improvement in force prediction accuracy over the baseline Allegro model, showcasing the potential of quantum-enhanced architectures for materials science applications.
5 The quantum layer employs a data re-uploading strategy that sequentially encodes features onto a limited qubit lattice with entangling gates, enabling complex nonlinear pattern recognition without requiring one qubit per feature.
6 Comprehensive quantum circuit analysis using ZX calculus revealed 100% parameter preservation after simplification, Fisher information metrics confirmed good trainability without barren plateaus, and Fourier analysis showed 79% expressivity with 127 non-zero coefficients out of 161 possible terms.
7 The Cu-Li dataset generation represents a significant contribution itself: 11,635 DFT-calculated structures including surface vacancies, adatoms, coherent interfaces, and melt-quench amorphized interfacial structures for modeling high-temperature Li-Cu material behavior.
8 Pareto front analysis revealed dataset-dependent trade-offs between accuracy and inference time, with Allegro MLP generally dominating on organic molecules while Allegro QDI showed particular strength on the inorganic metallic system.
💻Code: github.com/glatq/allegro
📜Paper: arxiv.org/abs/2602.16908
#MachineLearning #QuantumComputing #MaterialsScience #InteratomicPotentials #EquivariantNeuralNetworks #MultiObjectiveOptimization #DFT #MolecularDynamics #ComputationalChemistry #QuantumMachineLearning
5
1
9
1,246

23 Dec 2025
Long-range electrostatics for machine learning interatomic potentials is easier than we thought
1. This perspective article by Dongjin Kim and Bingqing Cheng introduces the Latent Ewald Summation (LES) framework, which can capture long-range interactions, charges, and electrical response in machine learning interatomic potentials (MLIPs) using only standard energy and force training data.
2. The authors distill two key design principles behind LES: (i) using a Coulomb functional form with environment-dependent charges to capture electrostatic interactions, and (ii) avoiding explicit training on ambiguous density functional theory (DFT) partial charges.
3. LES can be universally applied to augment any short-range MLIP with substantial flexibility. It allows for the inference or fine-tuning of dipoles and Born effective charges, and can incorporate charge/spin-state embeddings or tensorial targets.
4. The LES algorithm is simple and computationally efficient. It has been benchmarked on diverse systems, including bulk liquids, molecular crystals, polar molecules, ferroelectrics, and electrochemical interfaces, showing universal improvements in accuracy and reliable extraction of electrical response properties.
5. The article highlights that incorporating long-range electrostatics into MLIPs is simpler and more broadly applicable than commonly assumed. It also discusses current limitations and open challenges, such as extending LES to inhomogeneous systems and scaling to larger datasets.
6. The LES method is implemented in several MLIP frameworks, including MACE, NequIP, Allegro, CACE, CHGNet, and TACE. The implementations are publicly available, making it accessible for researchers to integrate LES into their own MLIP models.
📜Paper: arxiv.org/abs/2512.18029v1
💻Code: github.com/ChengUCB/les
#MachineLearning #InteratomicPotentials #LongRangeElectrostatics #MaterialsScience

7
894
20 Nov 2025
Fine-Tuning Unifies Foundational Machine-learned Interatomic Potential Architectures at Ab Initio Accuracy
1. A novel study demonstrates that fine-tuning can transform foundational machine-learned interatomic potentials (MLIPs) to achieve near-ab initio accuracy across diverse architectures. This work benchmarks five leading MLIP frameworks (MACE, GRACE, SevenNet, MatterSim, and ORB) on seven chemically diverse compounds, revealing dramatic improvements in force and energy predictions after fine-tuning.
2. Fine-tuning universally enhances force predictions by factors of 5-15 and improves energy accuracy by 2-4 orders of magnitude. This method effectively adapts general-purpose foundation models to capture system-specific interactions, enabling accurate predictions of atomistic and structural properties that are often inaccessible to short-timescale ab initio simulations.
3. The study introduces the aMACEing Toolkit, a unified interface for fine-tuning workflows across multiple MLIP frameworks. This toolkit simplifies the process by handling framework-specific complexities, allowing researchers to focus on scientific questions rather than implementation details. It also provides comprehensive analysis capabilities for trajectory post-processing.
4. The research highlights that fine-tuning is effective across both equivariant and invariant architectures, as well as conservative and non-conservative models. This universality suggests that the quality and relevance of training data are more critical than architectural details, offering flexibility in framework selection based on computational efficiency and user needs.
5. The study also evaluates the ability of fine-tuned models to reproduce key physical properties such as diffusion coefficients, radial distribution functions, and hydrogen bond dynamics. Fine-tuning enables accurate predictions of these properties, making MLIPs suitable for quantitative analysis and direct comparison with experimental data.
6. The findings suggest that fine-tuning should be a standard practice in molecular simulation workflows, providing a practical pathway to near-quantum chemical accuracy for extended simulations. The work paves the way for future development efforts focusing on inference speed and large-scale collaborative evaluation of MLIPs.
📜Paper: arxiv.org/abs/2511.05337v1
#MachineLearning #InteratomicPotentials #FineTuning #AbInitioAccuracy #MolecularDynamics #ComputationalChemistry

2
8
714
15 Oct 2025
Optimizing Cross-Domain Transfer for Universal Machine Learning Interatomic Potentials
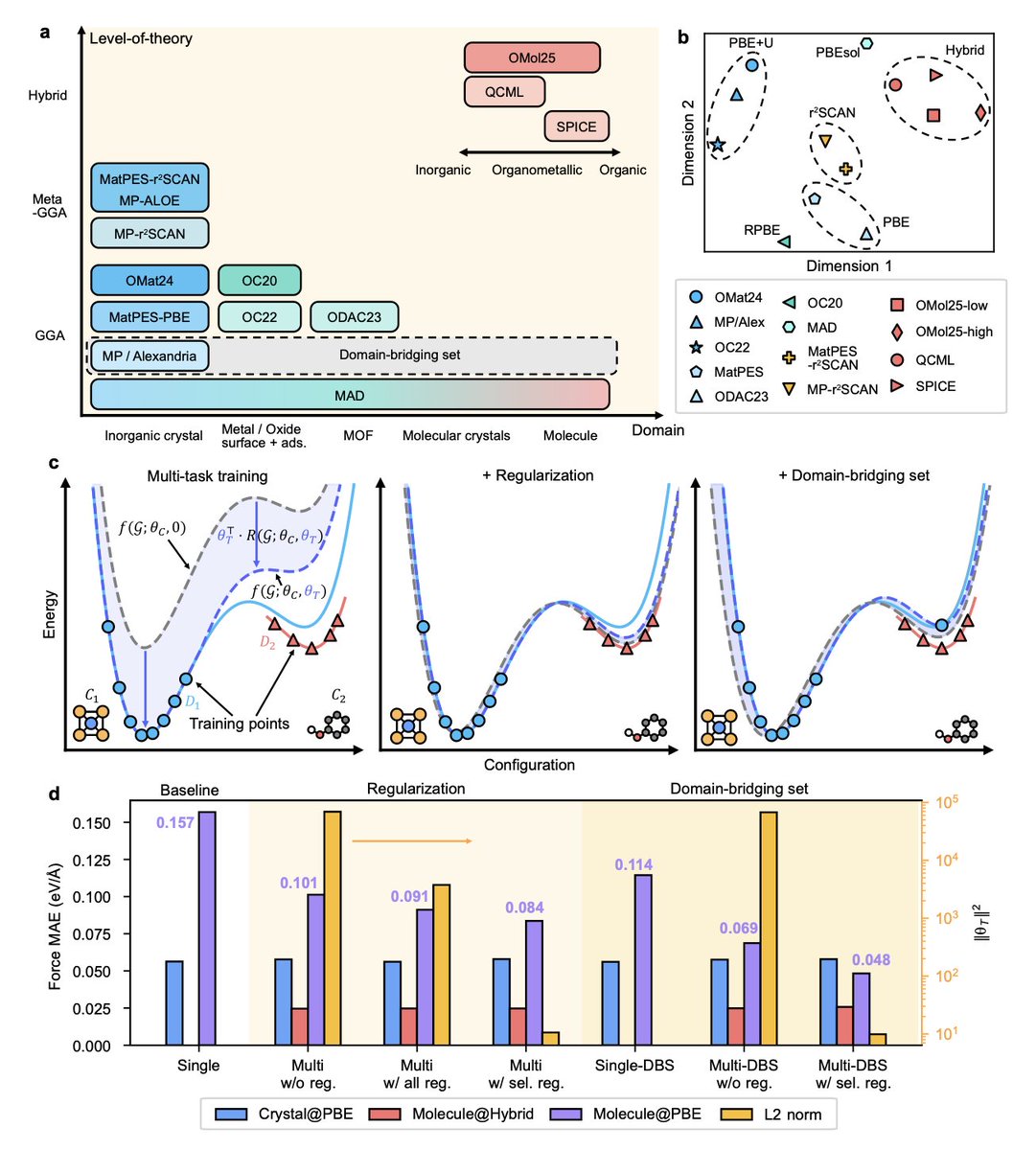
1. A new multi-domain training strategy for machine-learning interatomic potentials (MLIPs) is introduced, aiming to enhance their transferability across different chemical and functional domains. This is crucial for materials and chemical discovery, as most existing MLIPs overfit to narrow datasets or computational protocols.
2. The proposed method, SevenNet-Omni, incorporates selective regularization and a domain-bridging set (DBS) to align potential-energy surfaces across diverse datasets. This strategy significantly improves out-of-distribution generalization while maintaining high in-domain fidelity.
3. Trained on 15 open databases spanning molecules, crystals, and surfaces, SevenNet-Omni achieves state-of-the-art cross-domain accuracy. Notably, it demonstrates effective cross-functional transfer from large PBE datasets to reproduce high-fidelity r2SCAN energetics with only 0.5% r2SCAN data.
4. The model shows exceptional performance in various benchmarks, including adsorption-energy errors below 0.06 eV on metallic surfaces and 0.1 eV on metal-organic frameworks. This highlights its potential for applications in complex material systems.
5. The training strategy involves a curriculum learning approach, starting with inorganic bulk systems and progressively incorporating organic molecules and other datasets. This method ensures a balanced representation across different material domains.
6. The article also discusses the challenges of transferring knowledge across computational settings and chemical domains, especially between inorganic compounds and small molecules. The proposed framework effectively addresses these challenges, offering a scalable route toward universal, transferable MLIPs.
7. The performance of SevenNet-Omni is compared with other top-performing multi-task and single-task uMLIPs, consistently outperforming them in cross-domain scenarios. This includes applications in catalysis, molecular crystals, and hybrid organic-inorganic perovskites.
8. The study highlights the importance of accurate molecular energy predictions in determining overall model performance. The findings suggest that effective transfer learning can mitigate issues related to molecular energy overestimation and PES stiffening.
9. The article concludes that the proposed framework not only enhances the reliability of MLIPs across multiple domains but also provides a foundation for future developments in universal machine learning potentials for materials science.
📜Paper: arxiv.org/abs/2510.11241
#MachineLearning #MaterialsScience #InteratomicPotentials #CrossDomainTransfer #UniversalMLIPs
2
671
7 Oct 2025
Learning Inter-Atomic Potentials Without Explicit Equivariance
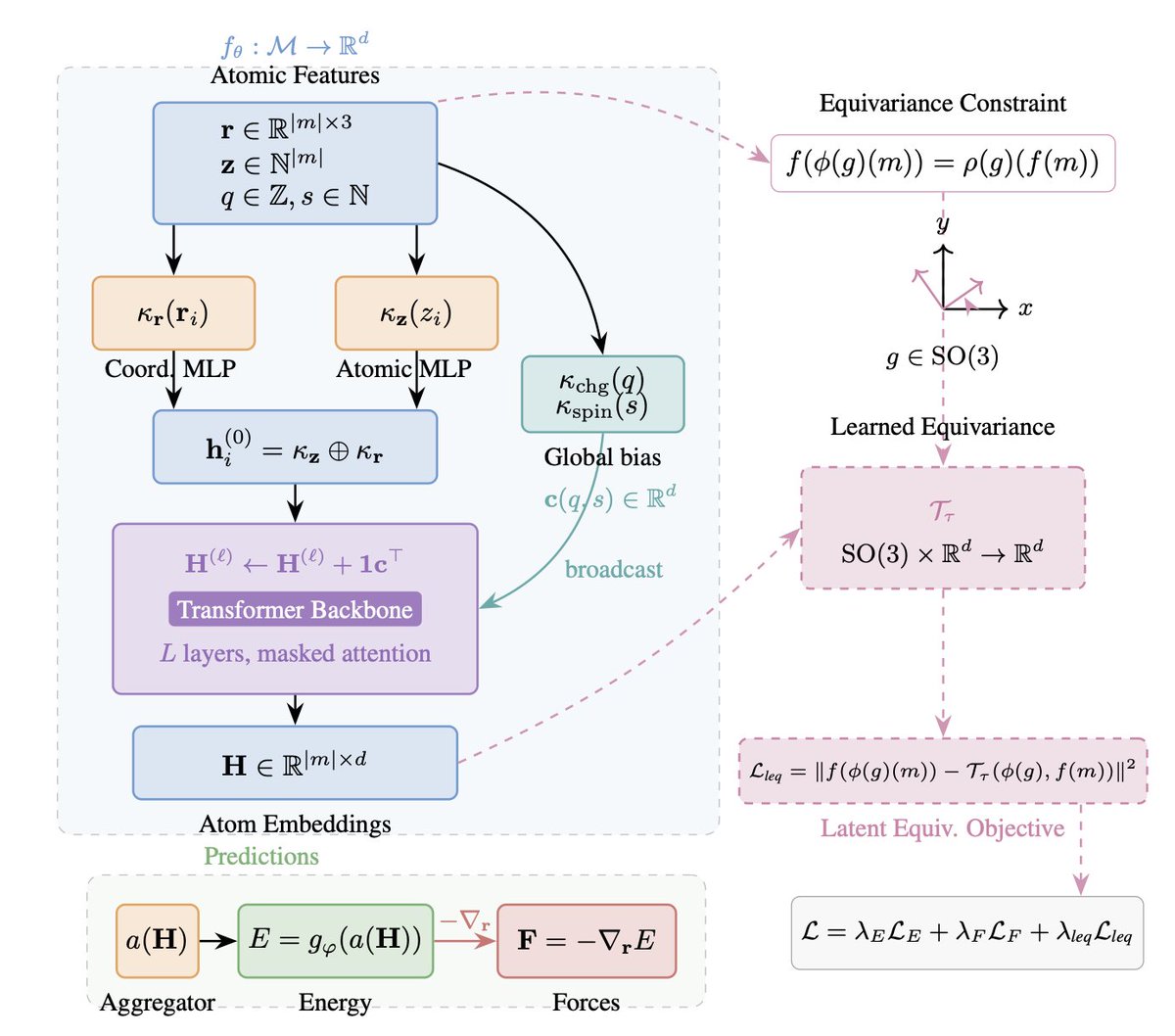
1. A new method called TransIP is introduced, which achieves molecular symmetry compliance in interatomic potentials without relying on explicit architectural constraints. This is a significant departure from current state-of-the-art models that enforce roto-translational symmetries through equivariant neural network architectures.
2. TransIP uses a Transformer-based model and incorporates a learned transformation network along with a contrastive objective to optimize for SO(3)-equivariance in the embedding space. This approach allows the model to retain the scalability and hardware efficiency of attention mechanisms while learning symmetry from data.
3. The study demonstrates that TransIP outperforms data augmentation techniques and achieves comparable performance to current state-of-the-art MLIP baselines on the Open Molecules 2025 (OMol25) dataset. This dataset is large and diverse, covering various types of molecules including small organics, biomolecular fragments, and electrolyte-like species.
4. The research shows that learned equivariance can be a powerful and efficient alternative to traditional equivariant or augmentation-based MLIPs. TransIP achieves 40% to 60% improvement in performance across varying OMol25 dataset sizes compared to a data augmentation baseline.
5. The training framework of TransIP includes an unconstrained Transformer backbone, a learned transformation network, and a contrastive objective for latent equivariance. The model is trained on a variety of molecular configurations, and the results indicate that the learned latent equivariance objective provides substantial improvements in force and energy predictions.
6. The authors suggest that the learned latent equivariance objective enables better performance scaling than popular data augmentation-based alternatives. This finding highlights the potential of using simpler and more scalable Transformer architectures to model equivariant molecular properties through learned symmetry.
📜Paper: arxiv.org/abs/2510.00027
#MachineLearning #MolecularSimulation #Transformer #Equivariance #InterAtomicPotentials
5
23
1,821
7 Oct 2025
Learning from the Electronic Structure of Molecules Across the Periodic Table
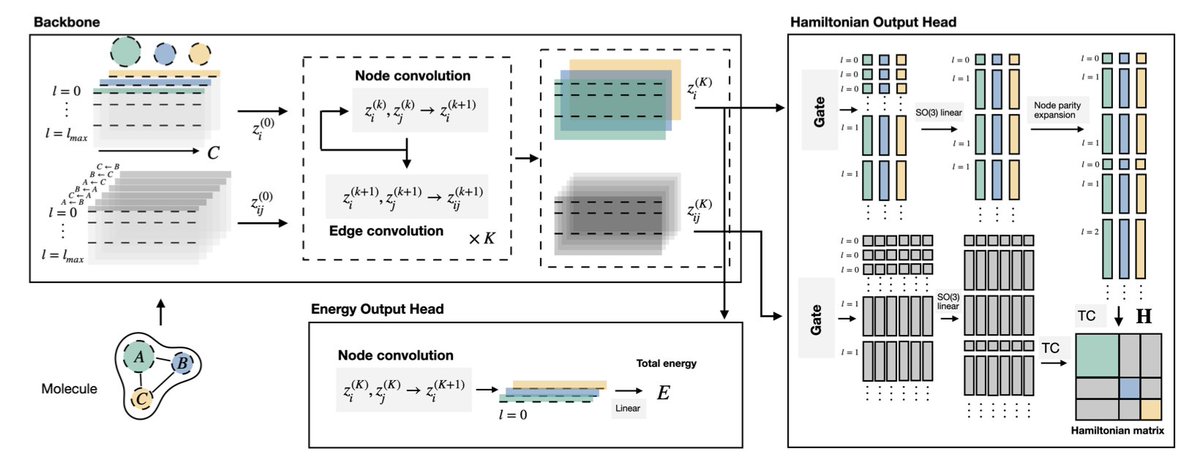
1. A new approach to training Machine-Learned Interatomic Potentials (MLIPs) has been introduced, leveraging the vast amounts of data contained in the Hamiltonian matrix to improve the prediction of atomic-level properties.
2. The authors developed HELM ("Hamiltonian-trained Electronic-structure Learning for Molecules"), a state-of-the-art model that bridges the gap between Hamiltonian prediction and universal MLIPs, scaling to structures with over 100 atoms and diverse elements.
3. A curated dataset called 'OMol_CSH_58k' was released, featuring unprecedented elemental diversity (58 elements), molecular size (up to 150 atoms), and basis set (def2-TZVPD), providing a rich source of data for training.
4. The concept of 'Hamiltonian pretraining' was introduced, where embeddings from Hamiltonian matrix training are used to improve energy prediction accuracy in low-data regimes, achieving up to a 2× improvement in controlled experiments.
5. The study highlights the potential of using electronic interactions as a transferable data source for representing chemical space, significantly enhancing the performance of MLIPs with limited training data.
6. The results demonstrate that HELM is state-of-the-art in Hamiltonian prediction benchmarks and that the learned features from Hamiltonian pretraining can be effectively repurposed for energy prediction tasks, improving data efficiency and generalizability.
📜Paper: arxiv.org/abs/2510.00224
#MachineLearning #Chemistry #ElectronicStructure #InteratomicPotentials #HamiltonianMatrix #DataEfficiency
1
4
14
1,022
30 Sep 2025
MLIP Arena: Advancing Fairness and Transparency in Machine Learning Interatomic Potentials via an Open, Accessible Benchmark Platform
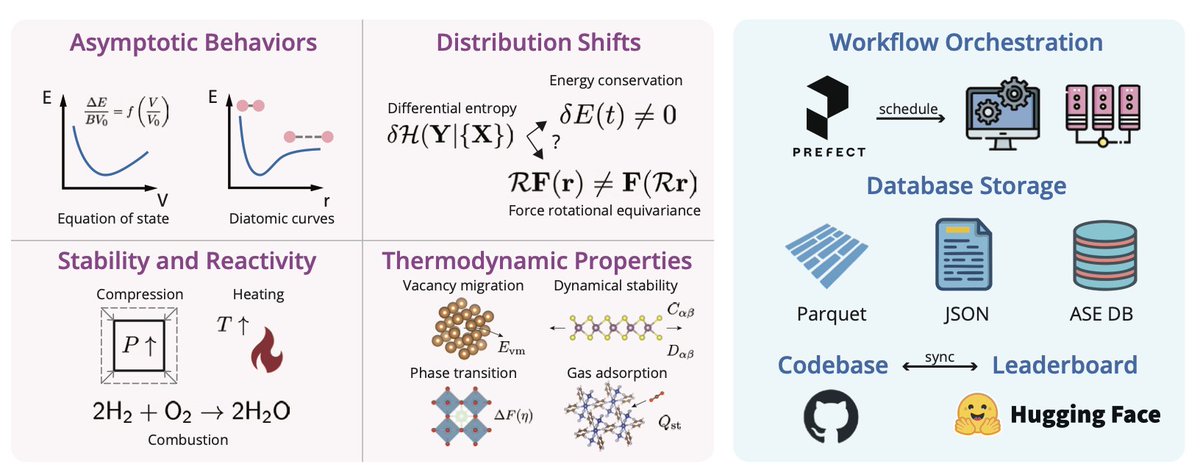
1. MLIP Arena introduces a comprehensive benchmark platform that evaluates machine learning interatomic potentials (MLIPs) beyond traditional error-based metrics. This platform focuses on assessing the physical soundness and practical utility of MLIPs in real-world applications.
2. The platform evaluates MLIPs across four primary perspectives: asymptotic behaviors, stability and reactivity, robustness to distribution shifts, and predictive capabilities for thermodynamic properties. This holistic approach provides deeper insights into the limitations and strengths of current MLIPs.
3. MLIP Arena proposes new metrics such as smoothness, short-range repulsion, and conservative field to quantify the quality of the potential energy surface (PES) and the physical laws critical for atomistic modeling. These metrics help in identifying models that better capture the correct dynamic interactions of atomistic systems.
4. The benchmark includes tasks like molecular dynamics simulations under extreme conditions, vacancy formation and migration, and CO2 adsorption in metal-organic frameworks. These tasks expose the robustness and reactivity of MLIPs in diverse scenarios.
5. MLIP Arena also evaluates the robustness of MLIPs to distribution shifts by monitoring energy conservation and rotational equivariance under varying local environments. This helps in understanding how well models generalize to out-of-distribution systems.
6. The platform provides a transparent and reproducible framework for developing the next generation of MLIPs with improved predictive accuracy and runtime efficiency while maintaining physical consistency. The Python package and online leaderboard are available for community use.
📜Paper: arxiv.org/abs/2509.20630v1
#MachineLearning #InteratomicPotentials #Benchmarking #MaterialsScience #ComputationalChemistry
1
3
5
1,125
26 Aug 2025
Graph Atomic Cluster Expansion for Foundational Machine Learning Interatomic Potentials
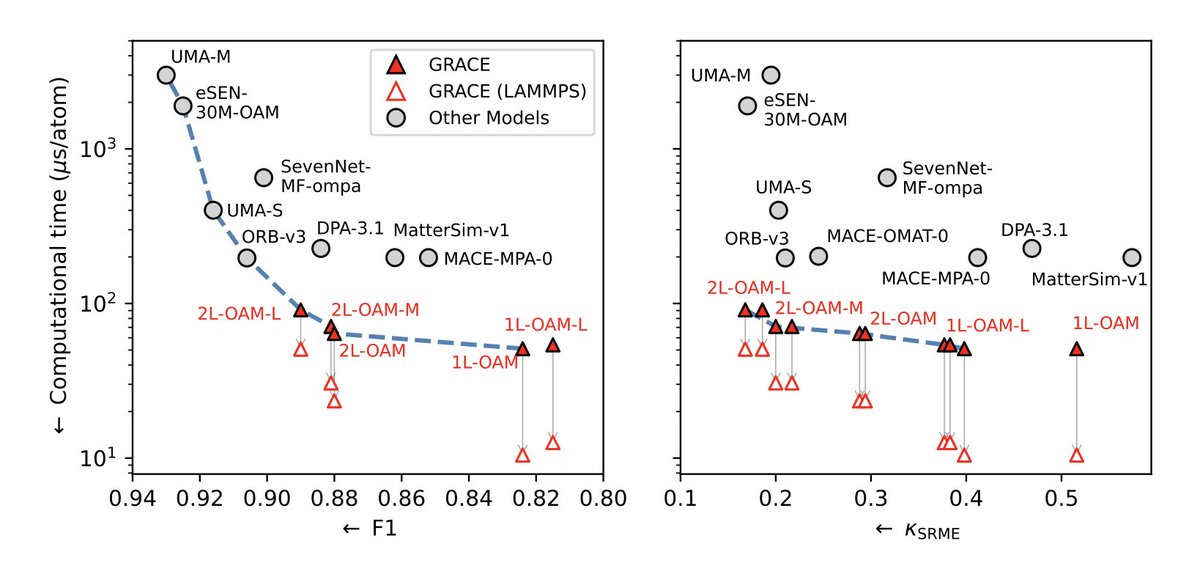
1. A novel study introduces GRACE models, a novel approach to machine learning interatomic potentials, trained on extensive materials datasets, offering unprecedented accuracy and efficiency across a wide range of materials.
2. GRACE models leverage a graph-based framework to capture complex atomic interactions, significantly outperforming existing methods in predicting material properties, with a superior balance of accuracy and computational speed.
3. The study demonstrates GRACE's exceptional versatility through fine-tuning and knowledge distillation, adapting the models to specialized tasks and simpler architectures while maintaining high accuracy and preventing catastrophic forgetting.
4. GRACE models achieve state-of-the-art performance in predicting thermal conductivity, a critical property for materials simulation, showcasing their robustness and ability to capture anharmonic contributions.
5. Comprehensive validation across diverse simulation tasks, including formation energies, elastic properties, and defect energies, confirms GRACE's effectiveness in describing equilibrium and non-equilibrium structures.
6. The study highlights GRACE's long-time stability in molecular dynamics simulations, accurately predicting dynamic properties such as radial distribution functions and diffusion coefficients over extended timescales.
7. Computational performance tests show that GRACE models deliver excellent efficiency, even on commodity GPUs, making them suitable for large-scale simulations with millions or billions of atoms.
8. Fine-tuning GRACE models on specialized datasets significantly improves their accuracy for specific tasks, such as Al-Li binary systems, outperforming models trained from scratch, especially in low-data regimes.
9. The study explores strategies to mitigate catastrophic forgetting during fine-tuning, demonstrating that freezing specific model layers can preserve general knowledge while learning new tasks.
10. Model distillation is successfully applied to create simpler, more computationally efficient GRACE models, achieving higher performance on a wider configurational space compared to models trained from scratch.
📜Paper: arxiv.org/abs/2508.17936
#MachineLearning #MaterialsScience #InteratomicPotentials #GRACEModels #ComputationalEfficiency #MaterialsDiscovery
1
1
4
827
29 Jul 2025
Iterative Pretraining Framework for Interatomic Potentials
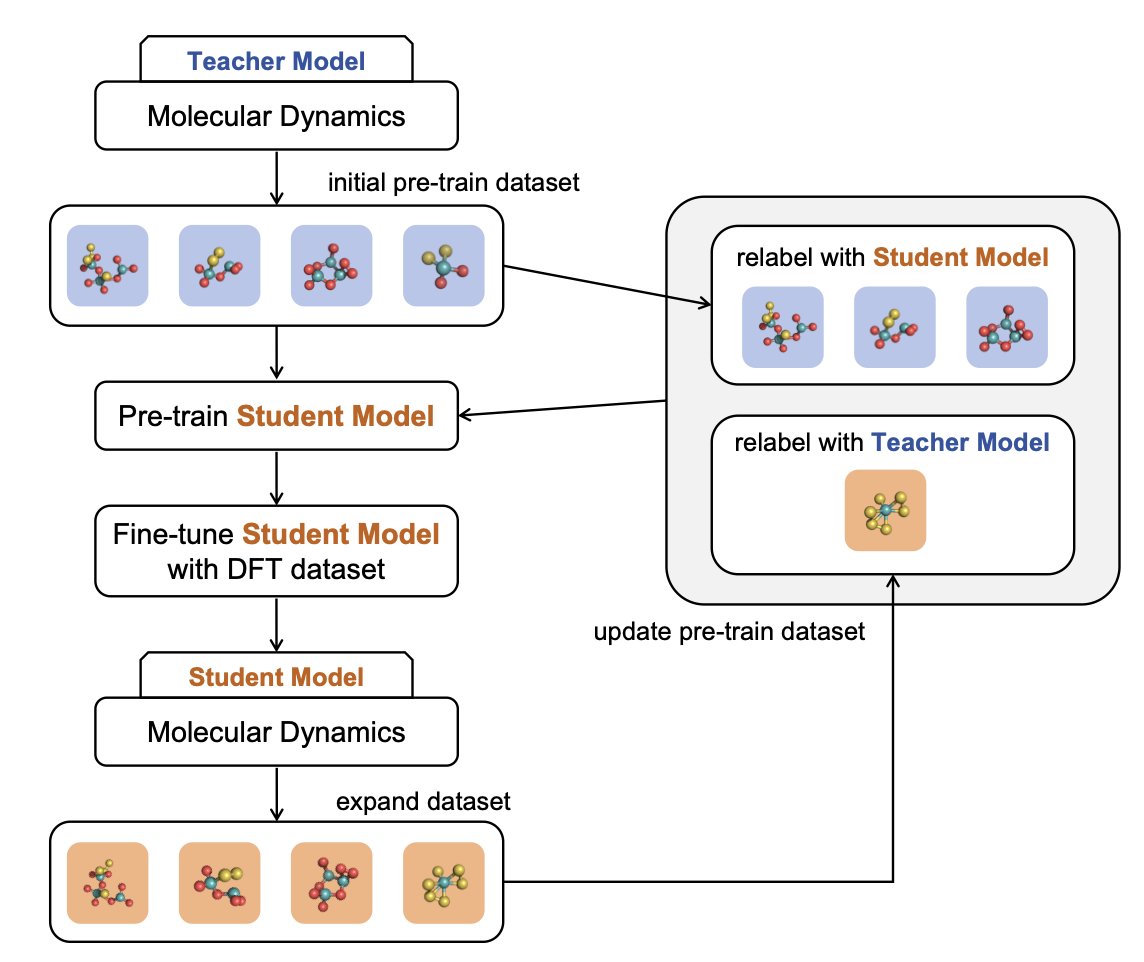
1. A new iterative pretraining framework called IPIP has been proposed to enhance the performance of machine learning interatomic potentials (MLIPs) for molecular dynamics simulations. This framework addresses the challenge of data scarcity and the mismatch between pretraining objectives and downstream tasks by iteratively improving model accuracy and efficiency without extensive labeled datasets.
2. IPIP incorporates a forgetting mechanism to avoid converging to suboptimal local minima during iterative training. It starts with MD simulations guided by a teacher model to generate an initial pretraining dataset, then fine-tunes a lightweight student model using high-quality data (e.g., DFT calculations). In subsequent iterations, the student model is used for simulations to expand the dataset, and pseudo-labeling strategies are employed to update the pretraining dataset.
3. The framework achieves significant improvements in accuracy and efficiency. Experiments on the MD-22 dataset and other complex systems (such as Chignolin and the Mo–S–O system) demonstrate that IPIP outperforms existing pretraining methods and general-purpose force fields. It reduces prediction errors by over 80% and speeds up simulations by up to 4× in the challenging Mo–S–O system.
4. IPIP’s iterative approach allows it to actively explore diverse regions in the configuration space through MD simulations, focusing on collecting edge conformations that are critical for improving model robustness. This self-enhancing training loop progressively improves the model’s accuracy, stability, and transferability across different atomic environments.
5. Future work may include integrating advanced uncertainty quantification techniques to guide data selection and enhance model reliability during iterative training. The method could also be extended to handle more complex chemical systems and coupled with active learning strategies to accelerate convergence and reduce computational cost.
📜Paper: arxiv.org/abs/2507.20118v1
#MachineLearning #MolecularDynamics #InteratomicPotentials #Pretraining #ComputationalChemistry
2
4
640
22 Jul 2025
A universal augmentation framework for long-range electrostatics in machine learning interatomic potentials
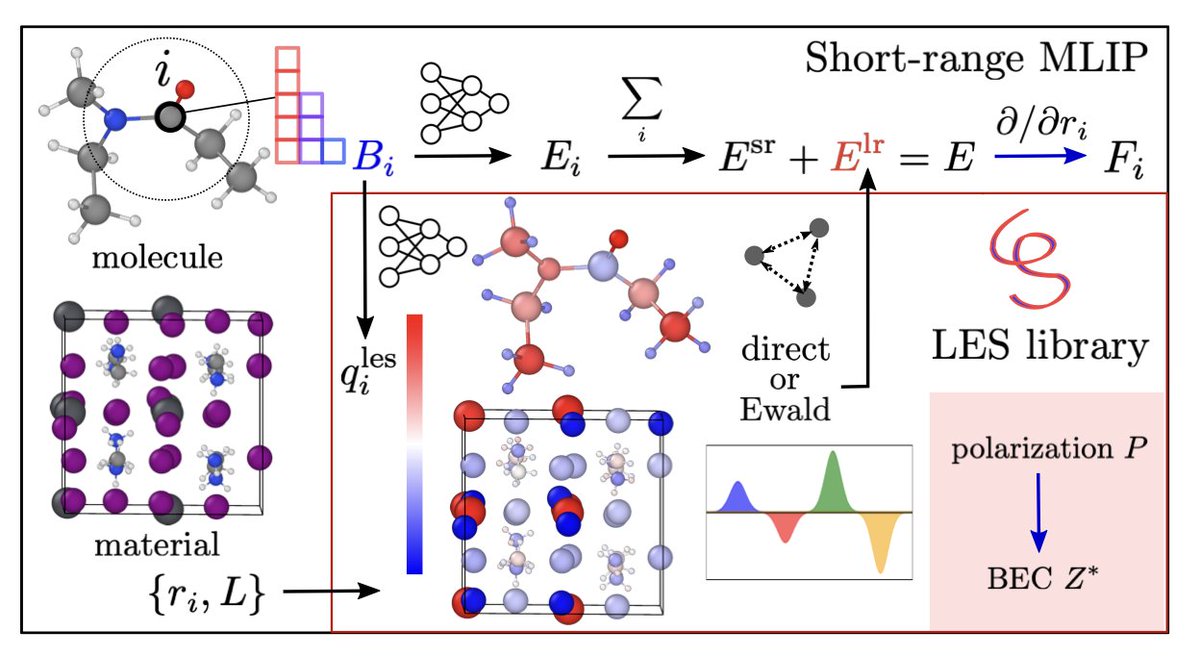
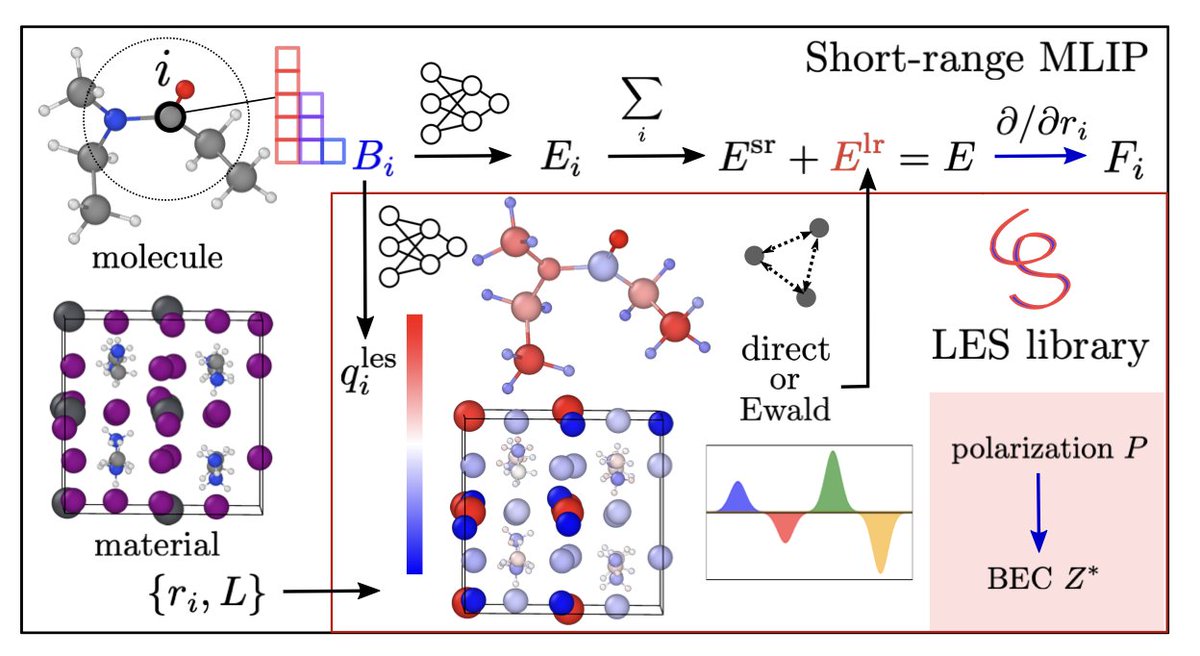
1. This novel study introduces the Latent Ewald Summation (LES) method, a universal framework to incorporate long-range electrostatics into machine learning interatomic potentials (MLIPs) without needing explicit charge labels. The LES method infers electrostatic interactions directly from energy and force data, significantly enhancing the accuracy of MLIPs for systems with significant electrostatics.
2. The LES framework is designed as a standalone library compatible with various short-range MLIPs, including MACE, NequIP, CACE, and CHGNet. It demonstrates remarkable improvements in predicting Born effective charges (BECs) and dipole moments, even when trained exclusively on energy and force data. This capability is crucial for accurately simulating systems with significant dielectric response, such as polar materials and charged molecules.
3. The study benchmarks LES-enhanced models on diverse systems, including bulk water, polar dipeptides, and gold dimer adsorption on defective substrates. Across all systems, LES not only reduces energy and force prediction errors but also accurately captures physical observables like BECs and adsorption energies. This highlights the robustness and versatility of the LES method.
4. The LES framework is scalable to large and chemically diverse datasets. The authors demonstrate this by training MACELES-OFF on the SPICE dataset, which includes organic molecules and biomolecules. MACELES-OFF outperforms its short-range counterpart in predicting bulk liquid properties and electrical response properties like IR spectra.
5. The LES method addresses a core limitation of current MLIPs by enabling efficient long-range electrostatics without additional training on electrical properties. This opens the door for developing universal MLIPs with full electrostatic physics, which can be applied across a wide range of chemical and biological systems.
💻Code: github.com/ChengUCB/les
📜Paper: arxiv.org/abs/2507.14302
#MachineLearning #Electrostatics #InteratomicPotentials #MaterialsScience #ComputationalChemistry #LongRangeInteractions
3
501
22 Jul 2025
A universal augmentation framework for long-range electrostatics in machine learning interatomic potentials
1. This novel study introduces the Latent Ewald Summation (LES) method, a universal framework to incorporate long-range electrostatics into machine learning interatomic potentials (MLIPs) without needing explicit charge labels. The LES method infers electrostatic interactions directly from energy and force data, significantly enhancing the accuracy of MLIPs for systems with significant electrostatics.
2. The LES framework is designed as a standalone library compatible with various short-range MLIPs, including MACE, NequIP, CACE, and CHGNet. It demonstrates remarkable improvements in predicting Born effective charges (BECs) and dipole moments, even when trained exclusively on energy and force data. This capability is crucial for accurately simulating systems with significant dielectric response, such as polar materials and charged molecules.
3. The study benchmarks LES-enhanced models on diverse systems, including bulk water, polar dipeptides, and gold dimer adsorption on defective substrates. Across all systems, LES not only reduces energy and force prediction errors but also accurately captures physical observables like BECs and adsorption energies. This highlights the robustness and versatility of the LES method.
4. The LES framework is scalable to large and chemically diverse datasets. The authors demonstrate this by training MACELES-OFF on the SPICE dataset, which includes organic molecules and biomolecules. MACELES-OFF outperforms its short-range counterpart in predicting bulk liquid properties and electrical response properties like IR spectra.
5. The LES method addresses a core limitation of current MLIPs by enabling efficient long-range electrostatics without additional training on electrical properties. This opens the door for developing universal MLIPs with full electrostatic physics, which can be applied across a wide range of chemical and biological systems.
💻Code: github.com/ChengUCB/les
📜Paper: arxiv.org/abs/2507.14302
#MachineLearning #Electrostatics #InteratomicPotentials #MaterialsScience #ComputationalChemistry #LongRangeInteractions
3
8
943
24 Jun 2025
Leveraging neural network interatomic potentials for a foundation model of chemistry
1.HackNIP is a hybrid framework combining pretrained neural interatomic potentials (NIPs) with shallow machine learning models. Instead of end-to-end deep learning, it extracts fixed-length embeddings from NIPs and feeds them into lightweight predictors for structure-to-property tasks.
2.This approach achieves state-of-the-art performance on 8 structure-property prediction tasks in the Matbench benchmark, outperforming both feature-based and deep neural network baselines in several cases.
3.HackNIP demonstrates exceptional data efficiency. On datasets with fewer than ~10⁴ samples, it often outperforms direct fine-tuning of the same NIP, making it ideal for limited-data regimes common in materials science.
4.The model remains competitive with larger end-to-end neural networks even at full dataset scale. On Matbench tasks with over 10⁴ samples, HackNIP shows performance within chemical accuracy and Pareto-optimal error margins.
5.HackNIP generalizes well beyond DFT-predicted properties. It achieves strong performance on diverse experimental and ab initio datasets, including Li-ion diffusivity, superconducting transition temperatures, and battery Coulombic efficiency.
6.It also shows robust classification performance on molecular-property prediction tasks from MoleculeNet (e.g., BBB penetration, toxicity), with AUC scores comparable to state-of-the-art molecular pretraining models despite using solid-material-trained NIPs.
7.A key innovation is identifying which NIP layer embeddings provide optimal features. Early layers often yield better predictions than deeper ones, especially for solid-state tasks, while molecular tasks benefit from deeper layers.
8.UMAP visualization and Jensen–Shannon divergence analyses show that early embeddings retain more task-relevant diversity. Late-layer embeddings become too compressed, losing fine-grained structural information crucial for downstream tasks.
9.Compared to end-to-end fine-tuning, HackNIP's simpler regressors undergo more uniform and efficient parameter updates, with less overfitting and better transferability, especially in small data regimes.
10.This modularity allows users to plug-and-play different NIP backbones and shallow learners. ORB MODNet yields the best results overall, but other combinations (e.g., MACE XGBoost) can be effective under different settings.
11.HackNIP lowers the entry barrier to high-performance materials ML. It is lightweight enough to run on standard hardware, robust across datasets, and suitable for practical use cases in both academia and industry.
12.This work suggests that hybrid approaches—assigning distinct roles to feature extraction and property mapping—can outperform monolithic models while improving interpretability and reducing computational cost.
💻Code: github.com/parkyjmit/HackNIP
📜Paper: arxiv.org/abs/2506.18497v1
#MachineLearning #MaterialsScience #TransferLearning #InteratomicPotentials #ML4Materials #ComputationalMaterials #GraphNeuralNetworks #NLPforScience

5
804

17 Jun 2025
Our new physics-informed method for training machine-learned #interatomicpotentials addresses atomistic simulation challenges by improving robustness & accuracy, reducing errors & minimizing data needs. Learn how in our #ICML2025 accepted paper: neclab.eu/research-groups/sy…. #NECLabs
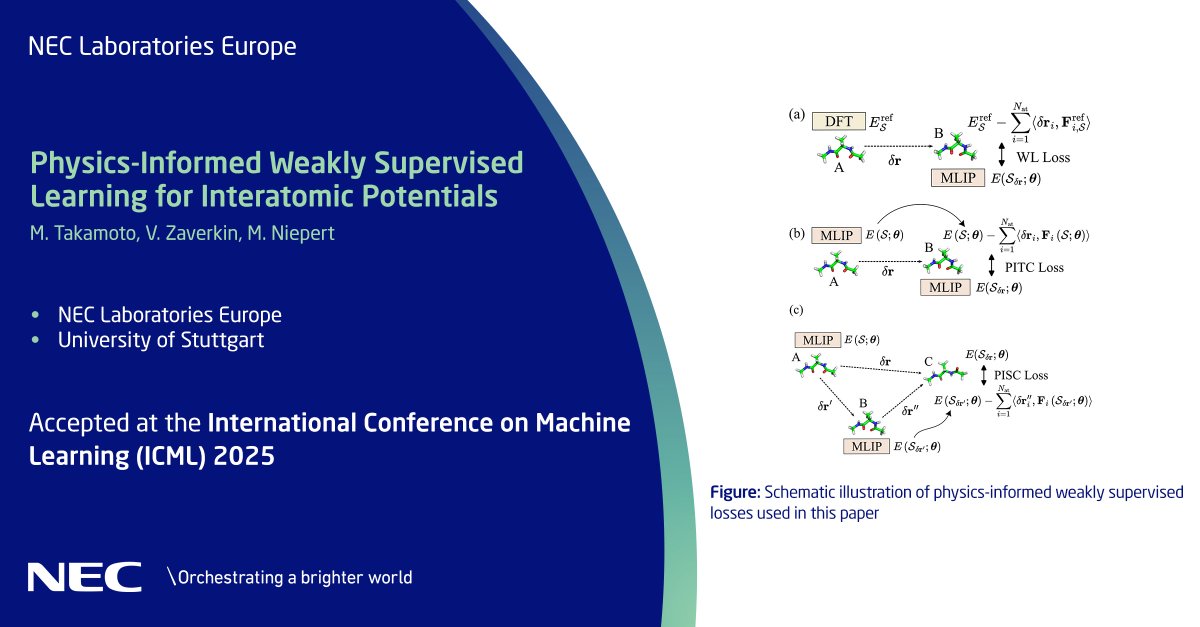
ALT Our new physics-informed method for training machine-learned #interatomicpotentials addresses atomistic simulation challenges by improving robustness & accuracy, reducing errors & minimizing data needs. Learn how in our #ICML2025 accepted paper: https://neclab.eu/research-groups/system-platform-for-iot-and-ai/intelligent-software-system/publications#publications-2143. #NECLabs
5
13
839

27 Mar 2025
Atomistic insights into iron and its oxides
open access: nature.com/articles/s41524-0…
@MPISusMat @maxplanckpress
#steel #alloys #theory #DFT #MD #simulation #greensteel #modeling #magnetic
#interatomicpotentials #metallurgy #iron #ore #oxide #thermodynamics #AI #machinelearning
5
162

11 Dec 2024
Join us today at #NeurIPS2024!
Higher-Rank Irreducible Cartesian Tensors for Equivariant Message Passing
🗓️ When: Wed, Dec 11, 11 a.m. – 2 p.m. PST
📍 Where: East Exhibit Hall A-C, Poster #4107
#MachineLearning #InteratomicPotentials #Equivariance #GraphNeuralNetworks
6 Dec 2024

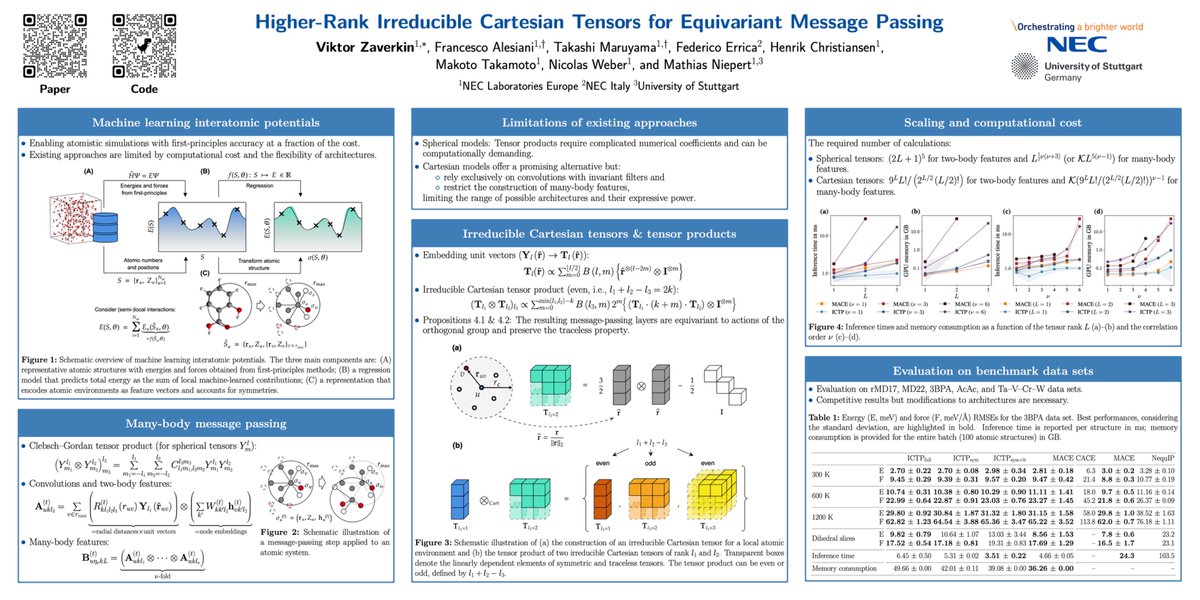
🚨Can we go beyond state-of-the-art message-passing models based on spherical tensors such as #MACE and #NequIP?
Our #NeurIPS2024 paper explores higher-rank irreducible Cartesian tensors to design equivariant #MLIPs.
Paper: arxiv.org/abs/2405.14253
Code: github.com/nec-research/ictp
1
12
528

30 Oct 2024
Paul Cuillier et al.: Integrating machine learning interatomic potentials with hybrid reverse Monte Carlo structure refinements in RMCProfile #ReverseMonteCarlo #MachineLearning #InteratomicPotentials @OhioState... #IUCr journals.iucr.org/paper?S160…
4
146

'Robust and scalable uncertainty estimation with conformal prediction for machine-learned #interatomicpotentials' by @zackulissi @medford_group @GTChBE @CMU_ChemE hits 1500 downloads! bit.ly/3V8Xlni #machinelearning #compchem #materials #AI #forcefields #quantum #DFT #HPC

3
11
1,977

Marcin Maździarz @ipptpan 🇵🇱 presents a systematic #quantitative comparative study of #silicon #interatomicpotentials for reproducing the properties of #silicene polymorphs:
👉 beilstein-journals.org/bjnan…
#DiamondOpenAccess 💎🔓#BJNANO
5
912
'Robust and scalable uncertainty estimation with conformal prediction for machine-learned #interatomicpotentials' by @zackulissi @medford_group @GTChBE @CMU_ChemE hits 1000 downloads!bit.ly/3V8Xlni #machinelearning #compchem #materials #AI #forcefields #quantum #DFT #HPC

1
6
1,343
'Robust and scalable uncertainty estimation with conformal prediction for machine-learned #interatomicpotentials' by @zackulissi @medford_group @GTChBE @CMU_ChemE hits 500 downloads! bit.ly/3V8Xlni #machinelearning #compchem #materials #AI #forcefields #quantum #DFT #HPC

2
4
841