PRISM: A High-Throughput Simulation Infrastructure for CADD Agents
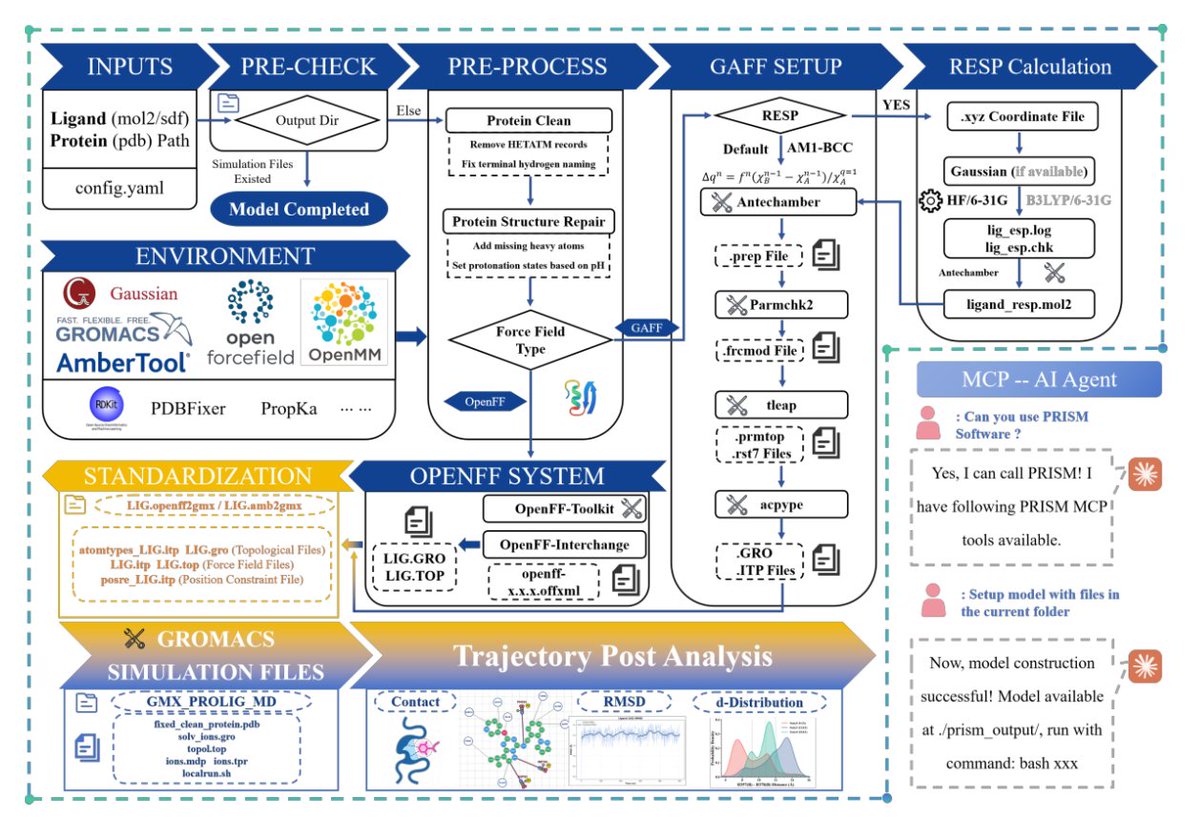
1. PRISM (Protein-Receptor Interaction Simulation Modeler) is presented as a GROMACS-native, Python-based infrastructure that turns fragmented protein–ligand MD workflows into a single, reproducible, high-throughput pipeline—designed to serve as a reliable backend for agent-driven CADD.
2. A key integration point is unified ligand parameterization behind one interface: GAFF/GAFF2 (AmberTools ACPYPE), OpenFF (SMIRNOFF via Interchange), CGenFF (stream parsing), OPLS-AA (LigParGen), and SwissParam options (MMFF/MATCH/hybrid). Outputs are standardized (GRO/ITP/atom types/restraints) so downstream steps are force-field-path agnostic.
3. For higher-quality electrostatics, PRISM adds an optional Gaussian RESP workflow (HF/6-31G* or B3LYP/6-31G*), allowing users to replace AM1-BCC charges while keeping the rest of the automated system build unchanged.
4. System construction is automated end-to-end: PDBFixer repairs structures (missing atoms/side chains/altloc issues), optional PROPKA assigns protonation states, pdb2gmx builds protein topologies, then PRISM merges ligand/protein topologies, solvates with configurable box shapes (default 1.5 nm padding), and sets ions (default 0.15 M NaCl).
5. Simulation control emphasizes reproducibility via a YAML configuration precedence system (CLI > user config > defaults) that generates/edits GROMACS .mdp files with validated defaults (PME, LINCS, v-rescale thermostat, stochastic cell rescaling barostat) and supports parallel multi-ligand directory organization (default production length reported as 500 ns).
6. Enhanced sampling is built in through automated REST2 setup: geometric temperature ladders (default 310–450 K), per-replica scaling rules (charges scaled by sqrt(λ), LJ ε by λ, etc.), per-replica topologies, and a single orchestration script—reducing the usual manual burden of replica workflows.
7. PRISM supports multi-tier binding energetics: endpoint MM/PB(GB)SA automation (gmx_MMPBSA or AMBER MMPBSA.py with topology conversion), in single-frame mode for fast triage or trajectory mode for averaging, with component decomposition (vdW, electrostatics, polar/nonpolar solvation).
8. The PMF module contributes a notable algorithmic piece: automated pulling direction optimization for umbrella sampling, using Metropolis–Hastings sampling on the unit sphere with simulated annealing to minimize steric hindrance (pocket-clearance mode or whole-protein collision mode), then auto-rotating the complex, elongating the box, generating SMD, extracting windows, and running WHAM.
9. PRISM-FEbuilder targets a common FEP pain point—hybrid topology construction—using distance-based atom mapping (default 0.6 Å) to classify atoms (common/transformed/surrounding) and manage charge differences with configurable strategies (reference-preserving, mutant-preserving, averaging). It emits GROMACS single-topology files with typeB/chargeB and dummy atoms, plus λ-window soft-core setup.
10. Results highlight two demonstrations: (i) an agent-orchestrated hierarchical screen on riboflavin synthase (ChEMBLFind → MolScope diversity selection → Vina docking → PRISM MM/PBSA) that not only recovers an active-site-like binder but also flags CHEMBL186010 binding at a trimerization-relevant C-terminal helix pocket, suggesting a potential allosteric/oligomerization-disruption site; (ii) FEbuilder benchmarking on HIF-2α, T4 lysozyme L99A, and p38α kinase with RMSE ~0.72–0.90 kcal/mol and generally small cycle-closure hysteresis.
💻Code: github.com/AIB001/PRISM
📜Paper: biorxiv.org/content/10.64898…
#MolecularDynamics #GROMACS #CADD #BindingFreeEnergy #FEP #UmbrellaSampling #REST2 #ForceFields #AIAgents #ComputationalChemistry #DrugDiscovery
10
1,601
Training a Force Field for Proteins and Small Molecules from Scratch
1 The authors identify a key limitation of conventional force fields: they are tuned manually for specific chemistries, which hampers transferability and makes systematic exploration of new functional forms difficult. To overcome this, they propose an end‑to‑end machine‑learning pipeline that learns all force‑field parameters directly from data.
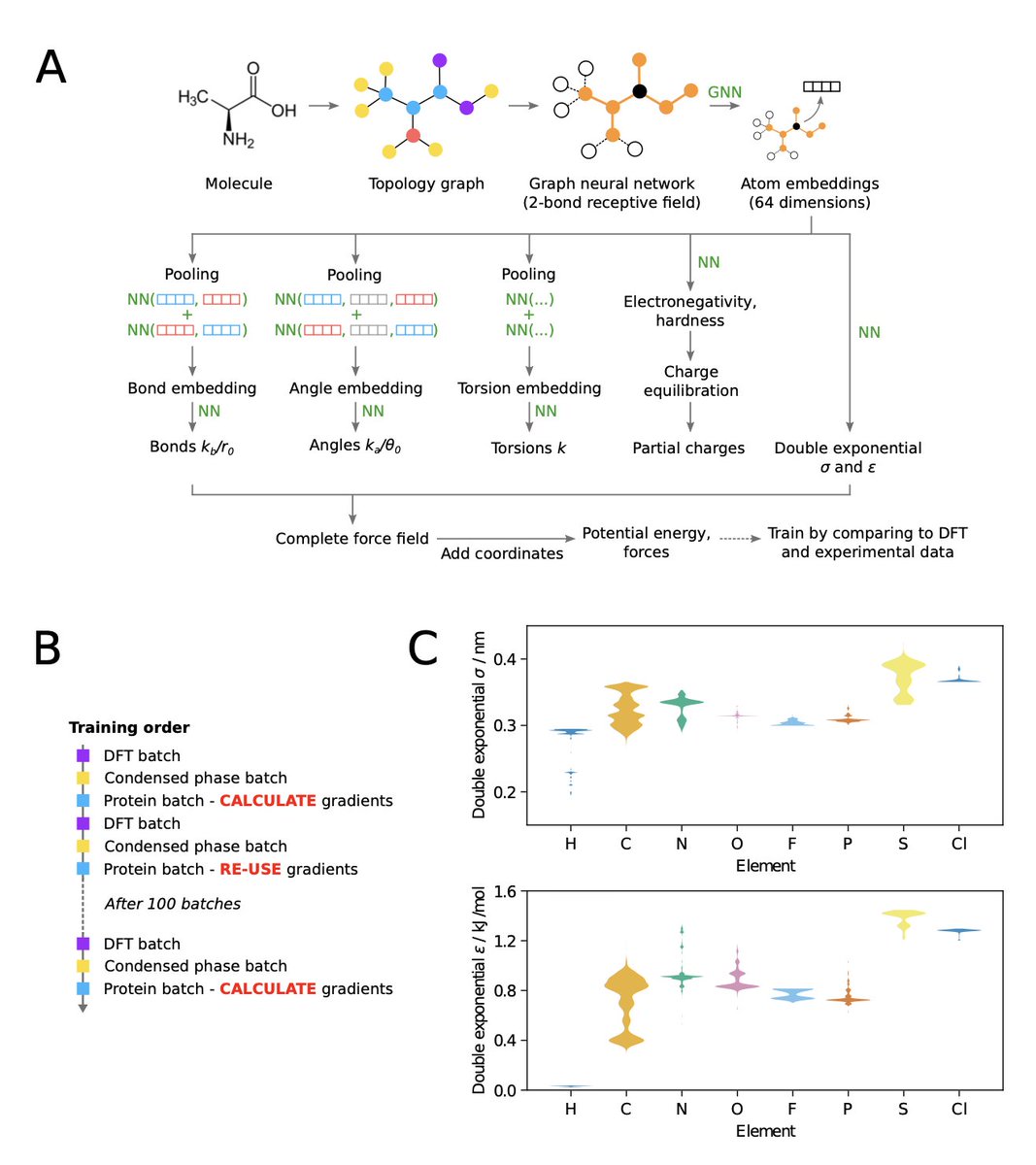
2 At the core is a graph neural network, Garnet, that assigns continuous atom types and predicts every bonded and non‑bonded parameter. Unlike prior work such as Espaloma, Garnet is trained from scratch—no legacy Lennard‑Jones or charge parameters are reused—using a blend of quantum‑mechanical forces, condensed‑phase thermodynamics, and protein NMR observables.
3 A major technical advance is the adoption of a double‑exponential potential for dispersion interactions. Training with the traditional Lennard‑Jones form proved unstable, whereas the double‑exponential, with two additional global parameters, converged smoothly and delivered performance on par with standard models, while remaining only marginally slower in OpenMM.
4 On small‑molecule benchmarks (OpenFF Industry Benchmark), Garnet reproduces QM‑optimized geometries and relative energies better than OpenFF 2.2.1 and comparable to Espaloma, while maintaining a low force‑field error on both bonded and non‑bonded terms. The model also captures torsional barriers with a TFD distribution similar to state‑of‑the‑art ML potentials.
5 Protein simulations of folded globular proteins (GB3, BPTI, HEWL, ubiquitin) remain stable for 5 µs and reproduce experimental scalar couplings with RMSEs comparable to Amber14SB and Espaloma. The model therefore demonstrates transferable accuracy across diverse biomolecular systems.
6 When applied to protein complexes, Garnet preserves the native interfaces over long trajectories, indicating that its intermolecular terms are sufficiently realistic. For intrinsically disordered proteins, the force field over‑compactifies the chains relative to a specialized IDP force field, yet it still captures the experimentally observed α‑helical propensity, suggesting room for targeted tuning.
7 The water model, trained without special treatment, reproduces TIP3P‑like density and dielectric constant but shows slightly over‑polarised oxygen charges due to the use of MBIS reference data. This highlights an avenue for future refinement of solvent parameters.
8 In relative binding‑free‑energy calculations using OpenFE, Garnet achieves a weighted RMSE of ~1.7 kcal mol⁻¹ across eight protein–ligand systems, matching the performance of the default OpenFE protocol and approaching that of commercial FEP . Kendall’s τ and fraction‑of‑best‑ligands metrics are similarly competitive, demonstrating that an automatically parameterised force field can support practical drug‑discovery workflows.
9 Extensive functional‑form exploration shows that the double‑exponential and buffered 14‑7 potentials train reliably, whereas the Lennard‑Jones and Buckingham forms introduce numerical instability. The study thereby provides a roadmap for future force‑field development that prioritises trainability alongside physical fidelity.
10 The authors argue that Garnet exemplifies a reproducible, automated pipeline for force‑field discovery that can be extended to nucleic acids, lipids, metals, and carbohydrates. By integrating new data and functional forms, the approach promises a universal, high‑accuracy classical force field that leverages machine learning without sacrificing speed.
💻Code: github.com/greener-group/gar… github.com/greener-group/ope…
📜Paper: arxiv.org/abs/2603.16770
#ComputationalBiology #MolecularDynamics #MachineLearning #ForceField #ProteinSimulations #DrugDiscovery #OpenScience
7
42
2,520

Feb 16
Merci ! 👍
Trop tard hélas.
Je suis retourné sur OpenFF pour compléter et la fiche produit a été complétée depuis hier.
Manifestement, ce produit fait parler de lui !
2
9

Feb 14
By saying that isoDDE is more accurate than FEP and TI RBFE with OpenFF
1
1
3
240

Jan 8
I spoiled myself this past holiday and started rewriting all of the open source comp bio tools that are in our stack because quite frankly I am sick and tired of a hundred dependencies every time I want to do a simple de novo molecule generation (and tired of conda too).
By the end of this spring, we will be dependency free. Everything and anything from molecule fingerprinting to dynamics to perturbation response scanning will be in one library. No more separate definitions of what a “topology” is between OpenMM, parmed, or openff-toolkit. Multithreading built in everywhere, by default, no more special functions to remember for RDKit. Batched MD simulations to take advantage of extra VRAM for protein ligand systems to run replicates all at the same time.
Oh and the library has a Python interface and compiles natively to WASM too because why not.
Multi-threading with inline assembly does wonders for Morgan fingerprint Tanimoto similarity - and this is only an M1 Max chip with only 10 cores.
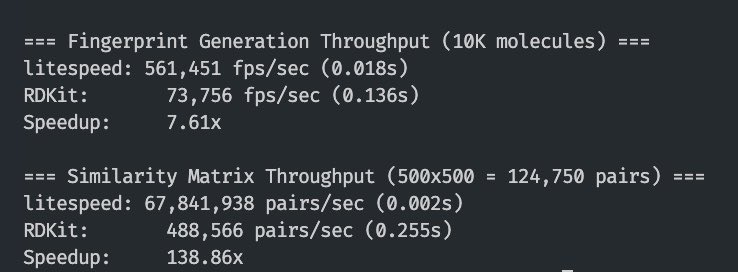
ALT Speed-up comparison vs RDKit
1
2
8
961

2 Nov 2025
Run MD simulation with Openff&Openmm on pixi's env #cheminformatics #RDKit #pixi #memo iwatobipen.wordpress.com/202…
3
5
36
3,869

3 Oct 2025
ABFE計算ソフトBindFlowが発表。無料オープンソースで、FEPやMM(PB/GB)SA法に対応。GROMACSを使用しGAFF、OpenFF、Espalomaをサポート
139組の複合体で検証した結果、既存の標準法と同等の精度を達成。MM(PB/GB)SA法はFEPに匹敵する相関を示しつつ計算コストを大幅に削減。
論文はリプ⬇️
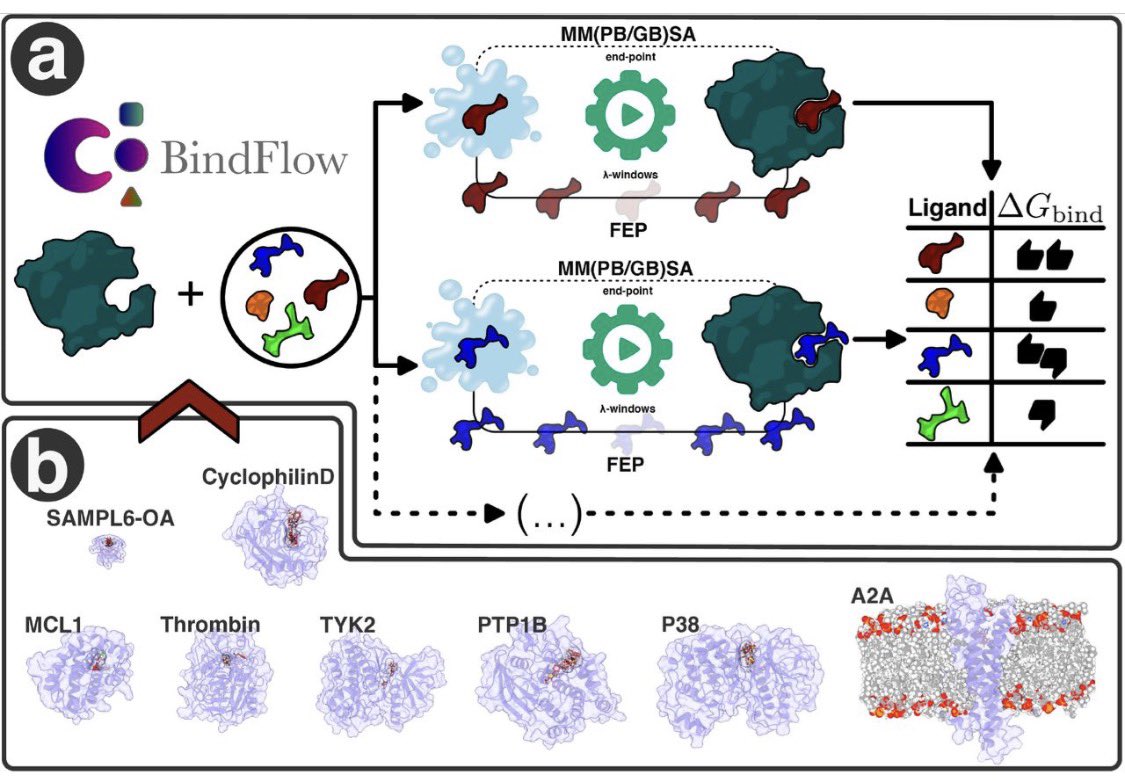
ALT Figure 1より引用(CC BY)
2
12
84
4,425

3 Oct 2025
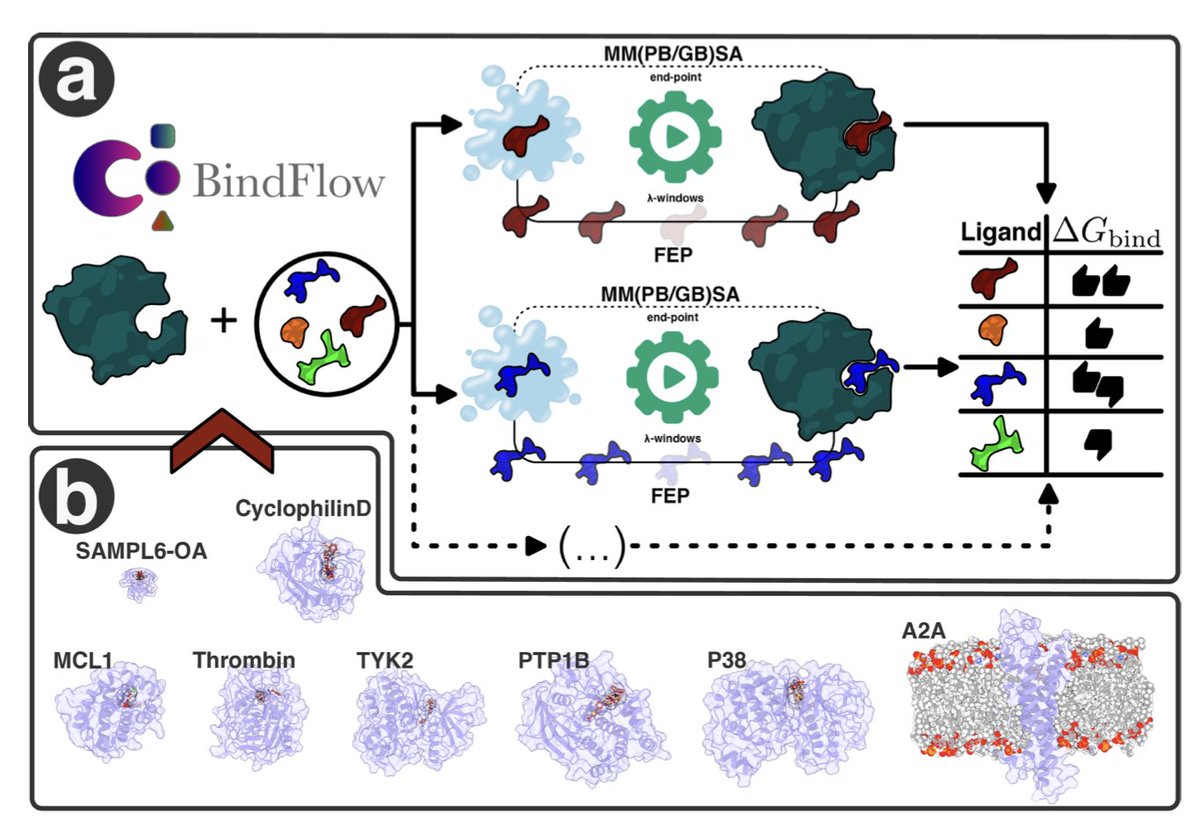
BindFlow: A Free, User-Friendly Pipeline for Absolute Binding Free Energy Calculations Using Free Energy Perturbation or MM(PB/GB)SA
1. BindFlow is a novel Python-based software pipeline designed for automated absolute binding free energy (ABFE) calculations, offering both free energy perturbation (FEP) and MM(PB/GB)SA methods. This tool aims to streamline the process of estimating binding affinities in drug discovery, making it more accessible and efficient.
2. The software is notable for its user-friendly design, extensive documentation, and tutorials, making it suitable for both beginners and advanced users. It supports multiple small-molecule force fields, including GAFF, OpenFF, and Espaloma, and can be run on various computing platforms, from workstations to distributed systems.
3. BindFlow was validated using a diverse set of 139 ligand/target pairs, encompassing eight different targets, including soluble proteins, a membrane protein, and a non-protein host–guest system. The results showed that BindFlow’s predictions are on par with established standards in the field, demonstrating its reliability and accuracy.
4. An intriguing finding is that MM(PB/GB)SA, despite being computationally less intensive than FEP, achieved correlations with experimental data that are comparable to FEP for some systems and force fields. This suggests that MM(PB/GB)SA could be a valuable tool for large-scale screening campaigns, balancing efficiency and accuracy.
5. The study highlights BindFlow’s potential to significantly enhance the efficiency and reproducibility of ABFE calculations, making it a practical platform for modern drug discovery. Its full automation and customization options make it a versatile tool for researchers.
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/ale94mleon/BindFl…
#BindFlow #ABFE #DrugDiscovery #MolecularDynamics #FreeEnergyPerturbation #MMPBSA #OpenSource #ComputationalBiology
2
23
122
5,818
13 Aug 2025
Bridging Quantum Mechanics to Organic Liquid Properties via a Universal Force Field
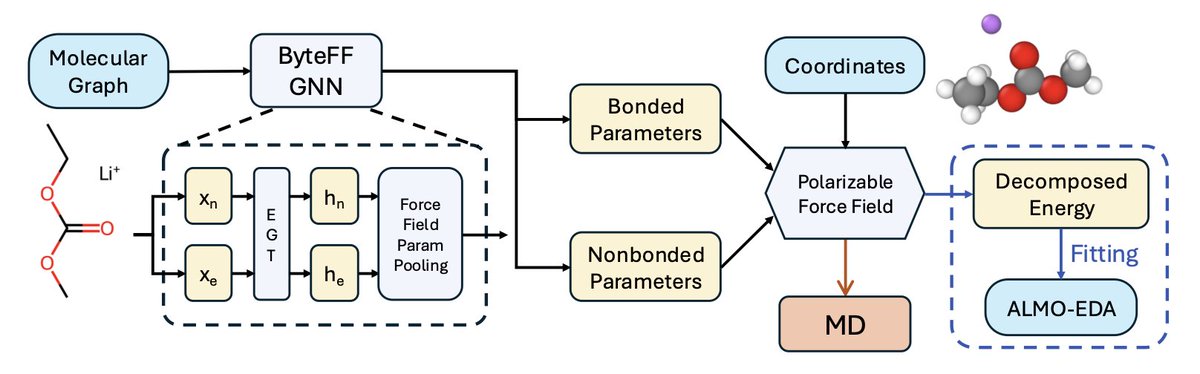
1. A novel study introduces ByteFF-Pol, a novel polarizable force field parameterized by a graph neural network (GNN) trained exclusively on high-level quantum mechanics (QM) data. This advancement bridges the gap between microscopic QM calculations and macroscopic liquid properties, offering a transformative tool for materials discovery.
2. ByteFF-Pol outperforms state-of-the-art classical and machine learning force fields in predicting thermodynamic and transport properties for small-molecule liquids and electrolytes. Its zero-shot prediction capability enables the exploration of previously intractable chemical spaces, making it a pivotal step toward data-driven materials design.
3. The study leverages physically-motivated force field forms and training strategies, allowing ByteFF-Pol to achieve exceptional accuracy without relying on experimental calibration. This is a significant departure from traditional force fields, which often require extensive experimental data for fine-tuning.
4. ByteFF-Pol's framework integrates molecular graphs with a GNN model to predict force field parameters directly. The energy function is partitioned into bonded and non-bonded components, with the non-bonded energy comprising repulsion, dispersion, permanent electrostatics, polarization, and charge transfer terms. This comprehensive decomposition aligns with the ALMO-EDA method, ensuring high accuracy.
5. The training dataset includes 60,790 unique molecular dimers with over 6 million conformations, covering a wide range of element types prevalent in organic molecules and electrolytes. The use of high-level DFT calculations for generating training labels ensures the model's robustness and transferability.
6. In benchmarks covering nearly 100 pure molecular liquids, ByteFF-Pol demonstrates remarkable accuracy in predicting densities and evaporation enthalpies, outperforming established force fields like OPLS-AA and OpenFF. The model's predictions are further validated through comparisons with experimental data for binary molecular mixtures and additional pure liquids.
7. ByteFF-Pol's applications in liquid electrolytes are particularly noteworthy. It accurately predicts density, viscosity, and conductivity across diverse electrolyte systems, demonstrating its potential for screening high-conductivity electrolytes and optimizing lithium salt concentrations. This capability is crucial for battery design and other electrochemical applications.
8. The study highlights ByteFF-Pol's potential for future optimization, particularly in enhancing its functional form to better model anisotropic interactions. This work represents a significant step toward ab initio-guided prediction of macroscopic properties, offering a scalable and data-efficient tool for materials discovery.
📜Paper: arxiv.org/abs/2508.08575
#QuantumMechanics #MolecularDynamics #ForceField #MachineLearning #MaterialsScience #Electrolytes #BatteryDesign
1
2
749

11 Aug 2025
Can't keep up with all the new computational tools? Here are the top ones to know for biology, chemistry and informatics
Bioinformatics
BLAST
MAFFT
Foldseek
MMseqs
Biotite
Biopython
Biopandas
RCSB PDB
UniProt
InterPro
𝘊𝘩𝘦𝘮𝘪𝘯𝘧𝘰𝘳𝘮𝘢𝘵𝘪𝘤𝘴 & 𝘴𝘮𝘢𝘭𝘭 𝘮𝘰𝘭𝘦𝘤𝘶𝘭𝘦𝘴
Chembl
Rdkit
Boltz-2
Pharmit (pharmacophore search)
AutoDock/GNINA
REINVENT
BindingDB
datamol & molfeat
GPCRdb
𝘈𝘯𝘵𝘪𝘣𝘰𝘥𝘺 𝘪𝘯𝘧𝘰𝘳𝘮𝘢𝘵𝘪𝘤𝘴
ANARCI
AbYsis
TAP
SabDab
PlabDab
AbodyBuilder/ImmuneBuilder
IgBlast
@tamarindbio Database & immune protein annotator
BioPhi
𝘗𝘳𝘰𝘵𝘦𝘪𝘯 & 𝘮𝘰𝘭𝘦𝘤𝘶𝘭𝘢𝘳 𝘥𝘦𝘴𝘪𝘨𝘯
AlphaFold2
Boltz-2
RFDiffusion
ProteinMPNN
PyRosetta
BindCraft
RFpeptides
Chai-1
𝘉𝘪𝘰𝘱𝘩𝘺𝘴𝘪𝘤𝘴 & 𝘥𝘺𝘯𝘢𝘮𝘪𝘤𝘴
GROMACS
OpenMM
OpenFE ( RBFE)
OpenFF
NAMD
AmberTools
PLUMED
Try out @tamarindbio to use (nearly) all of them, in a secure, scalable environment with no setup. required. On Tamarind, using our web interface or AI Copilot, you can connect all of the aforementioned tools together, deploy your custom tools, and create specialized versions for your own data.

9
107
600
32,880
17 Jul 2025
Deep Residual Learning for Molecular Force Fields
A new hybrid force field, ResFF, has been developed to enhance molecular simulations. It combines the strengths of traditional molecular mechanics force fields (MMFFs) with neural network force fields (NNFFs) by using deep residual learning.
ResFF shows superior accuracy in reproducing torsional profiles, a critical aspect for molecular simulations and drug discovery. It achieves an MAE of 0.45 kcal/mol on the TorsionNet-500 benchmark and 0.48 kcal/mol on the Torsion Scan benchmark, outperforming most contemporary models and approaching MACE's precision.
The force field also demonstrates exceptional precision in capturing intermolecular interactions, with an MAE of 0.32 kcal/mol on the S66×8 benchmark dataset. This capability is vital for accurately describing physical and biological processes like protein-ligand binding.
ResFF reliably reproduces energy minima on the OpenFF Industry Benchmark, consistently outperforming other methods in root-mean-square deviation (RMSD), torsion fingerprint deviation (TFD), and relative energy difference ($$$\Delta\Delta$$$E$). This indicates its ability to maintain equilibrium conformations and accurately predict relative stabilities.
In molecular dynamics simulations, ResFF enables efficient and robust performance. It explores small molecule conformational landscapes reliably over nanosecond-scale simulations, demonstrates polyalanine folding into a stable alpha-helix, and is applicable to heterogeneous protein-ligand systems like Tyk2, addressing the instability often seen in pure NNFFs.
The innovative three-stage training procedure for ResFF optimizes both its bonded and residual modules. This process ensures the force field integrates a robust physical foundation with data-driven corrections, effectively balancing accuracy and generalization.
💻Code: github.com/Ameki0/ResFF
📜Paper: doi.org/10.26434/chemrxiv-20…
#MolecularSimulation #DrugDiscovery #ForceFields #MachineLearning #ComputationalChemistry

15
892
25 Jun 2025
Condensation of Forcefield parameters from Machine Learning predicted statistical distributions for High-Throughput Virtual screening applications
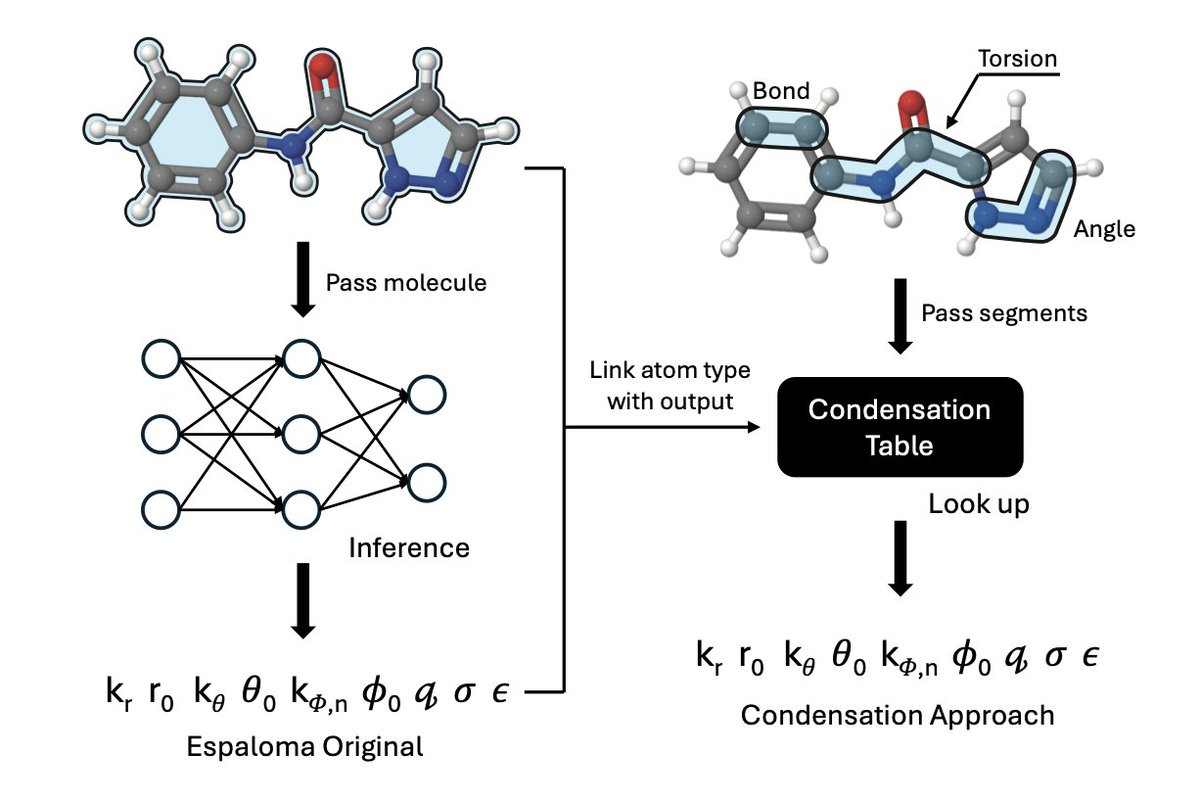
1.This work proposes a strategy to condense machine learning force field (MLFF) parameters into precomputed statistical tables, enabling 30x faster molecular structure generation without significantly sacrificing accuracy.
2.Traditional MLFFs like Espaloma are accurate but computationally expensive, making them incompatible with High-Throughput Virtual Screening (HTVS). The authors reverse-engineer MLFF outputs into lookup tables that mimic the speed of classical force fields like MMFF94.
3.Their condensed force field retains the flexibility of data-driven models but transforms prediction into a fast table lookup keyed on atom types, dramatically improving inference time during virtual screening.
4.The statistical condensation involves computing median parameter values (e.g., bond lengths, angles) across large datasets of Espaloma-predicted parameters, grouped by MMFF94 atom-type identifiers.
5.They benchmarked four force fields—Espaloma Class I, Condensed Espaloma, MMFF94, and Espaloma Class II—using Dompé’s LiGen platform, observing that Condensed Espaloma was the fastest and outperformed MMFF94 in both RMSD and TFD metrics.
6.Despite using simpler functional forms (Class I), Condensed Espaloma outperformed its Class II counterpart in predictive accuracy, largely due to optimization difficulties and ill-conditioning in Class II parameter fitting.
7.On the OpenFF Industry Benchmark Season 1 v1.1 dataset, Condensed Espaloma achieved comparable RMSD (0.618 Å) and TFD (0.066) to full Espaloma (0.542 Å RMSD, 0.055 TFD), while delivering a 30x speedup.
8.They also show that MLFF parameter assignment is a bottleneck, as neural inference is 9x slower than table-based retrieval. Their condensation framework removes this bottleneck entirely.
9.The authors retrained Espaloma models for both Class I and Class II functional forms, integrating the latter into LiGen for compatibility with MMFF94-style PES generation.
10.While Class II forms theoretically model PES more accurately, the lack of efficient optimization strategies for their parameter fitting leads to underperformance, making Class I more practical for now.
11.They note gaps in Espaloma’s training set—especially for torsional parameters—and propose active learning to expand its coverage and improve robustness in unseen chemistries.
12.Future work includes modeling non-covalent interactions more explicitly (e.g., pi-stacking, H-bonds) via specialized functional forms in Espaloma, leveraging its modular architecture.
13.The proposed condensation approach is scalable, interpretable, requires no inference at runtime, and matches transferable force fields in performance—while maintaining a pathway to continuous data-driven improvement.
💻Code: github.com/elvispolimi/force…
📜Paper: doi.org/10.26434/chemrxiv-20…
#Cheminformatics #MLFF #DrugDiscovery #VirtualScreening #MolecularMechanics #DeepLearning #ComputationalChemistry #OpenScience #ForceField #Espaloma #HighPerformanceComputing
19
1,187
25 Jun 2025
Condensation of Forcefield parameters from Machine Learning predicted statistical distributions for High-Throughput Virtual screening applications
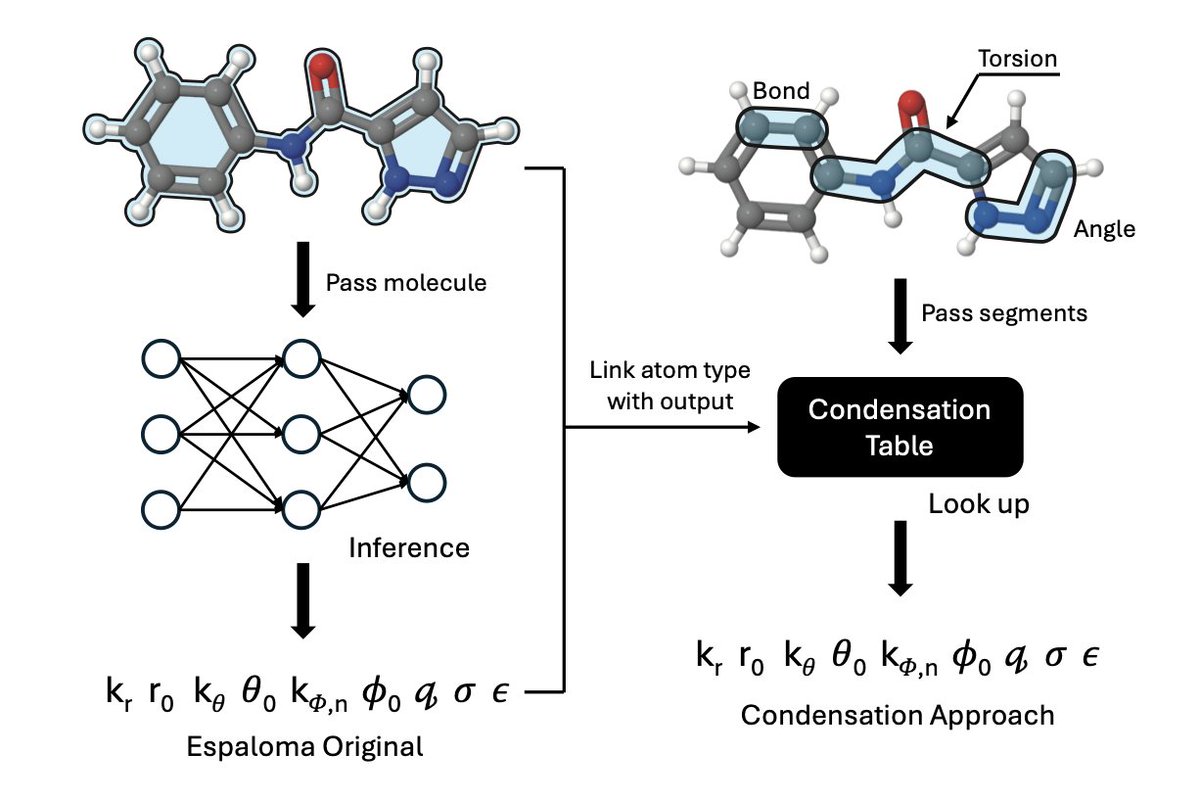
1.This work proposes a strategy to condense machine learning force field (MLFF) parameters into precomputed statistical tables, enabling 30x faster molecular structure generation without significantly sacrificing accuracy.
2.Traditional MLFFs like Espaloma are accurate but computationally expensive, making them incompatible with High-Throughput Virtual Screening (HTVS). The authors reverse-engineer MLFF outputs into lookup tables that mimic the speed of classical force fields like MMFF94.
3.Their condensed force field retains the flexibility of data-driven models but transforms prediction into a fast table lookup keyed on atom types, dramatically improving inference time during virtual screening.
4.The statistical condensation involves computing median parameter values (e.g., bond lengths, angles) across large datasets of Espaloma-predicted parameters, grouped by MMFF94 atom-type identifiers.
5.They benchmarked four force fields—Espaloma Class I, Condensed Espaloma, MMFF94, and Espaloma Class II—using Dompé’s LiGen platform, observing that Condensed Espaloma was the fastest and outperformed MMFF94 in both RMSD and TFD metrics.
6.Despite using simpler functional forms (Class I), Condensed Espaloma outperformed its Class II counterpart in predictive accuracy, largely due to optimization difficulties and ill-conditioning in Class II parameter fitting.
7.On the OpenFF Industry Benchmark Season 1 v1.1 dataset, Condensed Espaloma achieved comparable RMSD (0.618 Å) and TFD (0.066) to full Espaloma (0.542 Å RMSD, 0.055 TFD), while delivering a 30x speedup.
8.They also show that MLFF parameter assignment is a bottleneck, as neural inference is 9x slower than table-based retrieval. Their condensation framework removes this bottleneck entirely.
9.The authors retrained Espaloma models for both Class I and Class II functional forms, integrating the latter into LiGen for compatibility with MMFF94-style PES generation.
10.While Class II forms theoretically model PES more accurately, the lack of efficient optimization strategies for their parameter fitting leads to underperformance, making Class I more practical for now.
11.They note gaps in Espaloma’s training set—especially for torsional parameters—and propose active learning to expand its coverage and improve robustness in unseen chemistries.
12.Future work includes modeling non-covalent interactions more explicitly (e.g., pi-stacking, H-bonds) via specialized functional forms in Espaloma, leveraging its modular architecture.
13.The proposed condensation approach is scalable, interpretable, requires no inference at runtime, and matches transferable force fields in performance—while maintaining a pathway to continuous data-driven improvement.
💻Code:
github.com/elvispolimi/force…
📜Paper:
doi.org/10.26434/chemrxiv-20…
#Cheminformatics #MLFF #DrugDiscovery #VirtualScreening #MolecularMechanics #DeepLearning #ComputationalChemistry #OpenScience #ForceField #Espaloma #HighPerformanceComputing
1
3
646

16 May 2025
GUIすら開かず、クリック一つで出来るようにしたこと。
-フォースフィールドの自動設定(OpenFF)
-タンパク質とリガンドの自動マージ
-溶媒・イオン条件の自動設定
-温度・圧力制御(NVT/NPT)
-構造最適化・緩和 → 本番2ns
一時間半で出来るのでクラウドのGPUでたぶん50円くらいで出来ます。w
4
16
1,852
25 Mar 2025
5. 実行環境の信頼性
GPU (A100環境) を使用することで、CPUベースよりも高速かつ高精度なMDが可能です。
OpenMM OpenFFはエネルギー保存性に優れたアルゴリズムを採用しており、シミュレーション中の物理的整合性も保たれています。
1
10
2,432
25 Mar 2025
1. 標準プロトコルの遵守
本レポートでは、AutoDock Vina OpenMM OpenFF を用いたパイプラインを採用しています。これは多くのバイオインフォマティクス研究機関や製薬会社でも利用されている標準的かつ再現性の高い手法です。(名称の通りオープンソース)
1
1
9
255

4 Dec 2024
I'm not 100% sure either, but some scattered thoughts:
(1) AI2BMD is fast for an NNP but not yet to the speed that enables it to be a drop-in replacement for a FF in a classic MM workflow.
AI2BMD takes 2.6 s to evaluate 14K atoms. If we want to be able to do about 40 ns @ 4 fs timestep in 12 hours for e.g. FEP (1e7 force evals), each step needs to take about 5 ms, so we're still off by about 3 orders of magnitude. (This is still an issue with polarizable FFs like AMOEBA, which the authors compare to.)
There are lots of potential ways to get around this that @AriWagen discusses on the pod, but it's not as simple as "just use AI2BMD instead of OpenFF/OPLS4/whatever."

2
1
5
939

12 Jul 2024
Interested in what OpenFF is doing, but hard to see the big picture from our individual publications? We just published a review/perspective in J Phys Chem B that summarizes/highlights recent work! pubs.acs.org/doi/10.1021/acs…
40
116
14,221


14 Jun 2024
Nice, thank you corin! Re: setting starting coordinates. Openff molecules can be created from rdkit objects (with a 3D conformer) instead of smiles, although I dont know how that will work with packmol. Alternatively omm has an “AddSolvent” function
docs.openforcefield.org/proj…
1
4
511