
An electron-density point-cloud framework for robust protein-ligand interaction prediction
1. E-CloudBind reframes protein–ligand affinity prediction around electron-density point clouds rather than relying on sub-ångström atomic coordinates, aiming to stay accurate when structures are low-resolution or predicted (e.g., AlphaFold) and therefore noisy.
2. Key idea: replace hard distance cutoffs for “contacts” (e.g., within 5 Å) with density-aware Gaussian “electron clouds”, where interactions are defined by overlap/isosurface intersection, yielding a more resolution-agnostic interaction graph.
3. The framework explicitly splits chemistry into two complementary channels: non-covalent interactions from 3D electron-cloud point clouds, and covalent structure from intrinsic molecular graphs (bond topology), then fuses them for affinity regression.
4. Ligand electron density is obtained via semi-empirical quantum chemistry (GFN2-xTB), while protein pockets use a van der Waals radius-guided multivariate Gaussian sampling strategy as a physically motivated proxy that is far cheaper than full QM density.
5. Architecture highlights: K-means clusters point clouds into atom-aligned local regions; a point-cloud encoder (3D-GCN-style deformable kernels) learns local non-covalent patterns (e.g., H-bonds, π-stacking, van der Waals complementarity); a heterogeneous GNN encodes covalent graphs; a multi-bond fusion module integrates both.
6. On PDBbind 10-fold CV, E-CloudBind reports MAE 1.059 and Pearson 0.667, outperforming representative sequence-based (PSICHIC), graph-based (SIGN), and structure-based (DMFF) baselines, and also comparing favorably to recent structure-centric methods (EHIGN, Boltz-2, FlowDock) under the same protocol.
7. Robustness to experimental resolution: when regressing absolute error vs. crystallographic resolution, E-CloudBind shows a much flatter slope (0.017) than baselines (0.053–0.065), with a non-significant trend (p = 0.703), indicating reduced sensitivity to declining structural quality.
8. Robustness to structure source shifts: swapping experimental proteins with AlphaFold2 models causes only a small performance change for E-CloudBind (MAE 0.042; Pearson −0.004), while coordinate-dependent baselines degrade more (e.g., DMFF MAE 0.187; Pearson −0.093).
9. Out-of-distribution testing built from DAVIS via combinatorial partitioning by protein/ligand complexity shows tighter error dispersion for E-CloudBind (lowest median deviation), with stable performance across increasing protein Relative Contact Order and ligand Bertz complexity.
10. Practical and interpretability results: attention maps highlight polar ligand atoms and key pocket regions consistent with known interaction motifs; large-scale screening on 80,383 ZINC molecules against PBP1A, SARS-CoV-2 Mpro, and BCL-2 uses docking for follow-up, plus BCL-2 candidate assessment with synthesizability metrics and explicit-solvent MD (400 ns) suggesting stable binding for selected hits.
💻Code: github.com/Liuyujian0408/DPI ; doi.org/10.5281/zenodo.19851…
📜Paper: doi.org/10.1038/s41467-026-7…
#ComputationalBiology #DrugDiscovery #ProteinLigand #BindingAffinity #GeometricDeepLearning #GNN #PointCloud #ElectronDensity #VirtualScreening #AlphaFold

3
24
1,499

Jun 7
🧠 AI ΔΔG-Guided Discovery of Mutation-Resilient LRRK2 Inhibitors for Parkinson's Disease
A major challenge in Parkinson's disease (PD) drug discovery is that many kinase inhibitors lose potency against the pathogenic LRRK2 G2019S mutation, the most common genetic cause of familial PD.
A new study published in Journal of Computer-Aided Molecular Design demonstrates how integrating AI-based virtual screening, geometric deep learning, molecular docking, molecular dynamics, and ΔΔG mutation-resilience analysis can identify inhibitors that remain effective against both wild-type and mutant LRRK2.
The researchers screened 8,617 drug-like molecules using a multistage pipeline:
🔹 Uni-Mol Activity prediction
🔹 Schrödinger Glide docking
🔹 Uni-Mol BBB permeability prediction
🔹 DeepDock geometric rescoring
🔹 Rosetta Cartesian-ΔΔG mutation impact analysis
This workflow progressively reduced thousands of compounds to a small number of high-confidence candidates predicted to maintain binding after the G2019S mutation.
The standout hit was Compound 3.
Experimental validation revealed:
✅ WT LRRK2 IC50 = 14.21 nM
✅ G2019S LRRK2 IC50 = 14.75 nM
Remarkably, potency was essentially unchanged despite the disease-causing mutation.
Molecular dynamics simulations further showed stable binding throughout long-timescale simulations, with preservation of key hydrogen-bond interactions inside both WT and mutant kinase pockets. The favorable Rosetta ΔΔG score (-5.37) suggested that the mutation does not significantly destabilize ligand binding.
Another notable aspect is selectivity.
Compound 3 displayed minimal inhibition across a representative kinase panel including EGFR, PKCα, PIM1, TGFβ receptors, WEE1, ITK, and others, suggesting a promising starting point for further optimization.
Why this matters
The study is less about one specific inhibitor and more about a new paradigm:
Instead of screening only for potency against the wild-type protein, the authors incorporated mutation-aware ΔΔG calculations into the discovery workflow. This allows prioritization of compounds likely to remain active against clinically relevant disease variants.
As AI-driven drug discovery moves beyond simple affinity prediction, integrating structural biology, deep learning, and mutation-resilience modeling could significantly improve success rates for genetically defined diseases such as Parkinson's disease.
Reference
Ma Y, Yang X, Wang W, et al. Discovery of potent and selective LRRK2 inhibitors with preserved activity against the G2019S mutant via multi-stage virtual screening. Journal of Computer-Aided Molecular Design (2026). DOI: 10.1007/s10822-026-00840-3.
#ParkinsonsDisease #LRRK2 #DrugDiscovery #AIforScience #MachineLearning #ComputationalBiology #MedicinalChemistry #Neurodegeneration #VirtualScreening #DeepLearning #PrecisionMedicine #Biotech

1
3
146

Struct2Query: Structure-Guided Virtual Screening via Composite-Molecule ROCS Queries Derived from Protein Pocket Similarity #VirtualScreening
pubs.acs.org/doi/10.1021/acs…
@KShmilovich @VishnuSresht @genentech
#JCIM Vol66 Issue8 #compchem
7
24
1,307
Rise of AI Technologies in Virtual Screening #VirtualScreening #LLMs
pubs.acs.org/doi/10.1021/acs…
@hryhory_sinenka
#JCIM Vol66 Issue8 #Letter
7
24
1,065
MolViBench: Evaluating LLMs on Molecular Vibe Coding
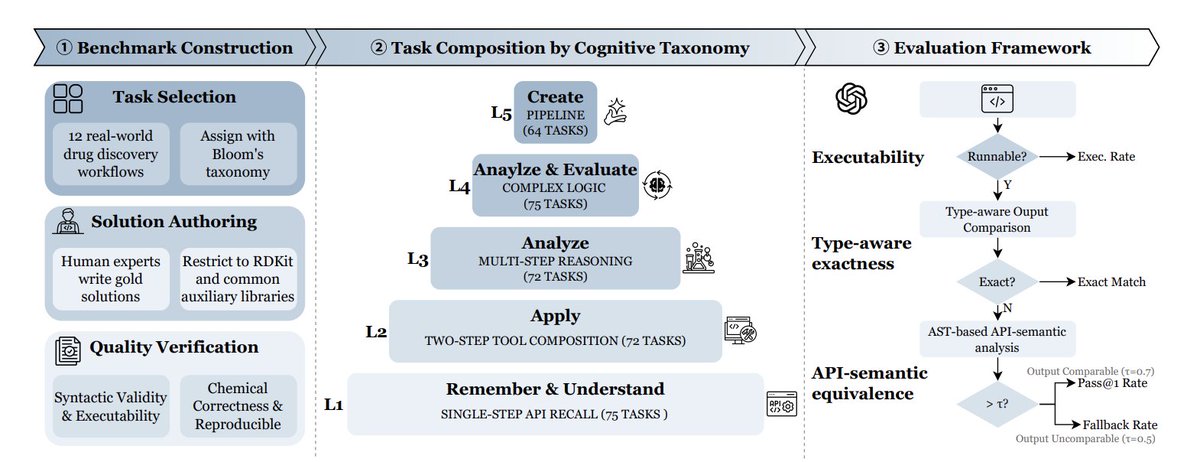
1 MolViBench is introduced as the first benchmark explicitly targeting “Molecular Vibe Coding”: chemists describe molecular tasks in natural language, and LLMs must generate executable RDKit-centric Python programs that are also chemically correct.
2 The benchmark contains 358 curated, real-world molecular coding tasks spanning 12 drug-discovery workflow categories (e.g., ADMET screening, virtual screening, reaction simulation, clustering/selection, QSAR/ML, stereochemistry, rule-based filtering, cheminformatics I/O), aiming to test the intersection of programming molecular understanding domain reasoning.
3 A key design choice is cognition-driven difficulty, organized into five Bloom-style levels: L1 single-API recall (descriptors, conversions), L2 two-step tool composition (fingerprints similarity, clustering, conformers), L3 multi-step chemical reasoning (reactions, ADMET heuristics, alerts like PAINS/Brenk), L4 branching/iteration/error recovery patterns, and L5 end-to-end pipeline creation (virtual screening pipelines, GA optimization, scaffold-split QSAR, Pareto/MPO ranking).
4 To make evaluation deterministic and comparable, tasks are constrained to RDKit as the primary toolkit (with limited auxiliary libs: numpy/pandas/scikit-learn/matplotlib/selfies). Each task has a human-authored gold reference solution under a unified interface (a single function named level_function), plus two-round quality verification for executability and chemical correctness.
5 The evaluation framework goes beyond plain unit-test exact match, because molecular tasks often have heterogeneous outputs and multiple valid solution paths. It uses a 3-stage scheme: (i) executability checks (compile entry-point runtime on multiple inputs with timeout), (ii) type-aware output comparison (tolerances for floats, canonical SMILES equivalence, recursive checks for structured outputs), and (iii) an AST-based API-semantic fallback that measures whether the generated code uses the appropriate RDKit functional categories when outputs are incomparable or when alternative valid implementations exist.
6 The paper reports systematic results on 9 frontier coding LLMs across three inference paradigms: Direct Generation (single shot), Incremental Repair (execute feed traceback for up to 3 repair rounds), and Agent Collaboration (Coder–Tester loop where a tester proposes test plans and validates plausibility).
7 Best overall performance is achieved by Claude Opus 4.6 Thinking with Incremental Repair (Pass@1 = 39.7%, Executable rate = 98.9%, Fallback Pass Rate = 72.6%). Across models, executability can be high while chemical/output correctness remains a bottleneck, motivating the multi-layer evaluation.
8 Performance drops monotonically with higher cognitive levels for all models; notably, every model remains below 10% Pass@1 on Level 5 tasks, highlighting a persistent gap in synthesizing long-horizon, end-to-end molecular discovery pipelines (planning, orchestration, and robust glue code).
9 Error analyses illustrate domain-specific failure modes: subtle RDKit API confusions that change return types (e.g., QED.qed vs QED.default), “over-engineering” outputs (returning dict metadata instead of requested scalar/SMILES), and chemically wrong reaction SMARTS due to small mapping/aromaticity constraints that yield fundamentally different products.
10 Incremental Repair is found to be consistently robust across models and levels, while Agent Collaboration is more sensitive: it can help weaker models but may degrade stronger models by introducing unnecessary rewrites that break partially correct solutions.
💻Code: github.com/phenixace/MolViBe…
📜Paper: arxiv.org/abs/2605.02351
#LLM #CodeGeneration #Cheminformatics #RDKit #DrugDiscovery #Benchmark #MolecularAI #VirtualScreening #QSAR #ADMET
3
759
Improving AlphaFold2 Performance in Virtual Screens Targeting GPCRs by Enhancing Binding-Site Conformational Sampling
1. The paper introduces AFsample2T, a targeted AlphaFold2 sampling strategy that boosts virtual screening performance for GPCRs by generating diverse binding-site conformations rather than relying on a single “best” structure.
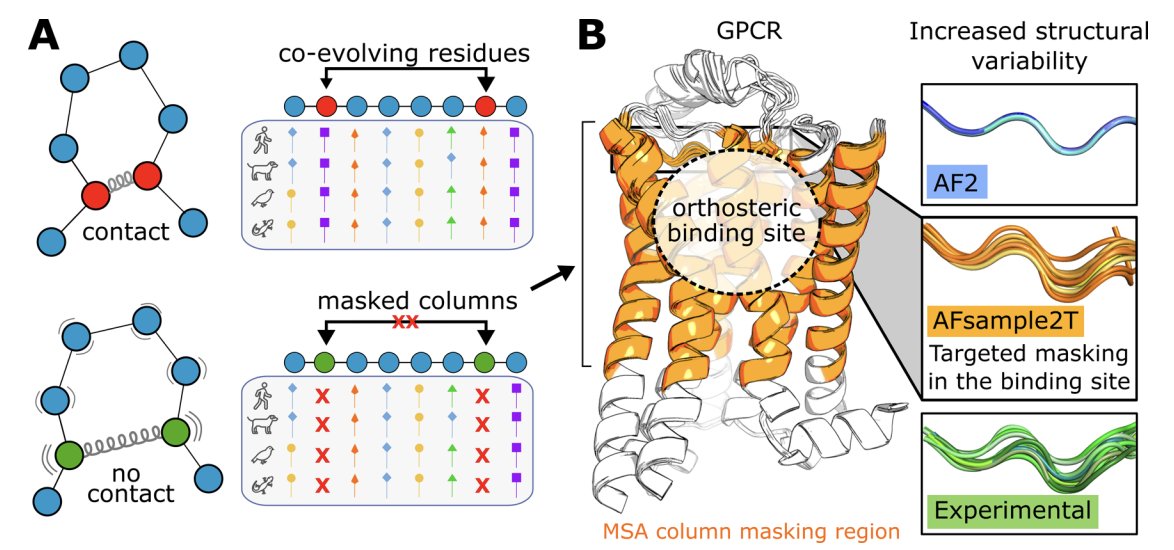
2. Core idea: mask selected MSA columns only in the orthosteric-site region (extracellular-facing TM segments part of EL2) to weaken local coevolution constraints and encourage alternative pocket geometries, while keeping the rest of the receptor well-constrained.
3. AFsample2T contrasts with global masking (AFsample2), which reduced binding-site accuracy for GPCRs here (AUC 0.38). Targeted masking preserved fold quality while improving pocket modeling, showing that “where to perturb” matters as much as “how much to perturb”.
4. The authors benchmarked 10 class A GPCRs using 61 curated experimental binding-site structures (from an initial pool of 119 PDB structures) and generated 1,000 models per receptor to quantify how often predicted pockets match experimental ones within 1–2 Å side-chain RMSD (symmetry-aware).
5. Moderate targeted masking improved binding-site accuracy: default AF2 AUC 0.54; adding dropout without masking AUC 0.57; targeted masking at 10–30% reached AUC ~0.59–0.61. Too much masking (50%) degraded secondary structure and collapsed performance (AUC 0.43).
6. The best-performing ensemble (AFsample2T) mixes 250 models each from 0%, 10%, 20%, and 30% masking (with dropout), yielding AUC 0.63 and capturing 73.8% of experimental binding sites at 1.5 Å RMSD vs 60.7% for AF2 (a 22% relative gain at that threshold).
7. A key mechanistic improvement is realistic binding-site plasticity. Median binding-site side-chain RMSF increased from 0.15 Å (AF2; overly rigid pockets) to 0.45 Å (AFsample2T), approaching experimental variability (median 0.58 Å). Backbone RMSF similarly moved from 0.10 Å (AF2) to 0.28 Å (AFsample2T), close to experiment (0.30 Å).
8. AFsample2T also mitigates a known docking issue: AF2 often predicts narrow/collapsed pockets. Across receptors, mean pocket volume increased (209 → 218 Å3) and the “top 1% most open” pockets expanded substantially (272 → 389 Å3), closer to experimental pockets (mean 256 Å3). This was especially relevant for μ-opioid receptor, where AF2 pockets were too collapsed.
9. Virtual screening evaluation used DOCK3.8 with ChEMBL actives (52–202 per receptor) and property-matched ZINC20 decoys, totaling extremely large-scale docking (reported as >240 trillion complexes scored). Rigid-receptor docking was used, making pocket microstates critical.
10. Ensemble screening ligand-guided model selection is the practical win: while median enrichment of AF2-based models remained below experimental structures, the top 1% AFsample2T models improved early enrichment (mean aLogAUC top 1%: 10.8 → 12.9; mean EF1% top 1%: 7.5 → 9.6). In some targets (e.g., TAAR1, μ-opioid receptor), best AFsample2T models approached top experimental-structure performance.
11. The paper provides a workflow for prospective use: generate ≥250 AFsample2T models for the relevant receptor state, dock a ligand/decoy control set to compute enrichment, select ~top 1% models, manually inspect key interactions/poses, then proceed to large-library prospective screening.
12. Modeling receptor state is handled explicitly: inactive models use receptor sequence alone; active models are generated by cofolding receptor with heterotrimeric G protein sequences via AF2-Multimer, capturing hallmark TM6 movements and separating “state sampling” from “pocket microstate sampling”.
💻Code: github.com/wallnerlab/AFsamp…
📜Paper: doi.org/10.1021/acs.jcim.6c0…
#AlphaFold2 #GPCR #VirtualScreening #Docking #StructureBasedDrugDesign #ComputationalChemistry #Bioinformatics #ProteinStructure #DrugDiscovery #MachineLearning
15
90
4,981
MolViBench: Evaluating LLMs on Molecular Vibe Coding
1. MolViBench frames a practical gap: “Molecular Vibe Coding,” where chemists describe molecular tasks in natural language and expect LLMs to generate executable RDKit-based Python that is both runnable and chemically correct—beyond tool-calling agents and beyond text-only chemistry QA.
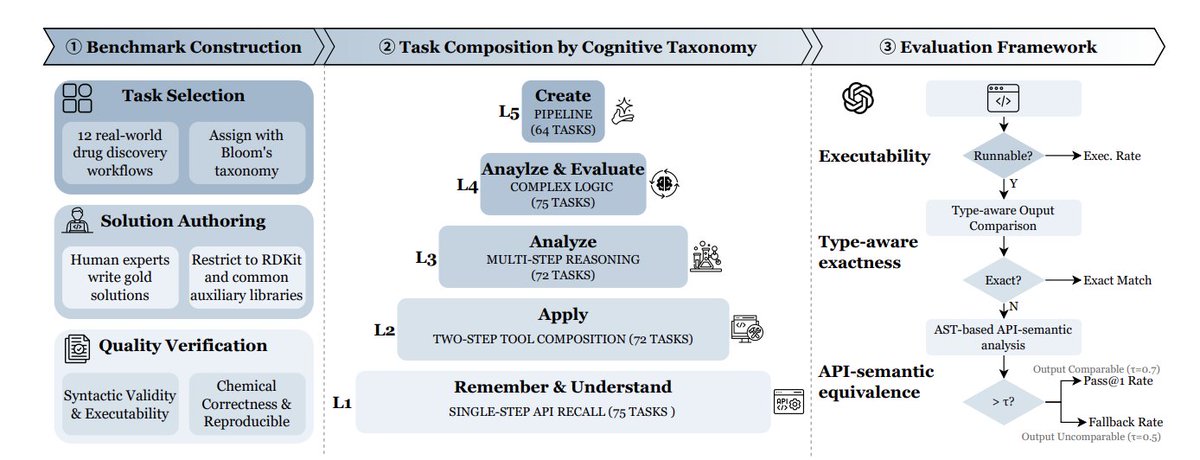
2. The benchmark contains 358 curated, expert-authored tasks spanning 12 real drug-discovery workflows (e.g., molecular characterization, ADMET screening, virtual screening, combinatorial chemistry, lead optimization, reaction simulation, clustering/selection, QSAR/ML, stereochemistry, rule-based filtering, and cheminformatics I/O).
3. A key design choice is cognition-driven difficulty, organized into 5 levels aligned with Bloom’s taxonomy: L1 single-step API recall; L2 two-step tool composition; L3 multi-step chemical reasoning (reactions, ADMET logic, alerts); L4 branching/iteration/error recovery patterns; L5 creating end-to-end pipelines (screening, optimization, QSAR with scaffold split, Pareto ranking).
4. MolViBench restricts implementations to RDKit (plus numpy/pandas/scikit-learn/matplotlib/selfies) to reduce cross-toolkit nondeterminism, and provides gold reference solutions under a unified interface (a single function named level_function), with two-round quality verification and standardized test molecule sets.
5. The paper argues that executability alone is insufficient in chemistry: code can run yet be chemically wrong (e.g., stereochemistry mishandling, incorrect reaction SMARTS, invalid products). MolViBench therefore evaluates both “can it run?” and “is it chemically/semantically right?”
6. The evaluation framework is multi-layered: (i) executability checks (compile, entrypoint existence, runtime on multiple inputs with timeout), (ii) type-aware output comparison (tolerances for floats, canonical SMILES equivalence, recursive checks for structured outputs), and (iii) an AST-based API-semantic fallback that measures overlap between predicted vs. reference RDKit API categories when exact output comparison is infeasible or overly strict.
7. Reporting goes beyond Exact Match: the main metric Pass@1 triggers API-semantic fallback only when outputs are structurally incomparable (using a coverage threshold τ=0.5), while a broader “Fallback Pass Rate” can credit executable solutions that miss exact outputs but match API semantics strongly (τ=0.7), highlighting partial correctness and format divergence.
8. The study benchmarks 9 frontier LLMs and compares three real usage paradigms: Direct Generation (single shot), Incremental Repair (execute feed traceback for up to 3 fixes), and Agent Collaboration (Coder–Tester loop with independent test-plan generation and iterative feedback).
9. Main results show a consistent ceiling for current models on real molecular workflows: best overall is Claude Opus 4.6 Thinking with Incremental Repair (Pass@1 39.7%, executable rate 98.9%, Fallback Pass Rate 72.6%). Performance drops monotonically from L1 to L5, and all models remain under 10% Pass@1 on Level 5 pipeline-creation tasks, suggesting long-horizon orchestration is the core bottleneck.
10. Paradigm insights: Incremental Repair is the most consistently helpful across models/levels, while Agent Collaboration can help weaker models but may degrade stronger ones due to unnecessary rewrites that disrupt partially correct solutions—especially on higher cognitive levels where preserving intermediate structure matters.
💻Code: github.com/phenixace/MolViBe…
📜Paper: arxiv.org/abs/2605.02351
#LLM #Cheminformatics #RDKit #Benchmark #CodeGeneration #DrugDiscovery #VirtualScreening #ADMET #QSAR #ScientificML
1
12
1,170
LiBRe: A Ligand-Aware Sequence-Based Binding Residue Prediction Model for Virtual Screening #VirtualScreening
pubs.acs.org/doi/10.1021/acs…
@GiltaeSong
#JCIM Vol66 Issue7 #MachineLearning #DeepLearning
3
24
979
Advancing Ligand-based Virtual Screening and Molecular Generation with Pretrained Molecular Embedding Distance
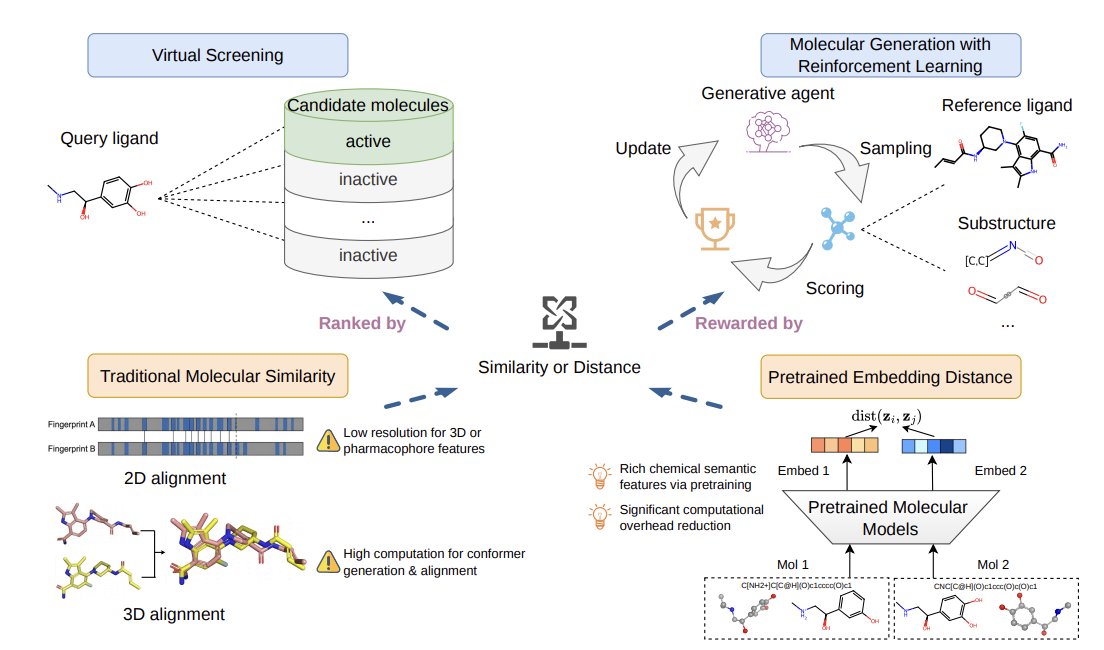
1 PED (Pretrained Embedding Distance) is introduced as a task-agnostic molecular similarity signal computed directly from frozen pretrained molecular models—no similarity labels, no target-specific finetuning, and no conformer alignment required.
2 The paper instantiates PED with two very different foundation models: GeoDiff (dual encoder with 2D GIN 3D SchNet, pretrained for conformation generation) and MoLFormer (SMILES Transformer pretrained on 1.1B molecules). Distances are computed in embedding space (cosine or Euclidean), optionally using 2D-only, 3D-only, or concatenated 2D 3D embeddings.
3 A key question addressed: do pretrained embeddings implicitly encode the same information that classic 3D similarity tools measure (shape pharmacophore/color), even when the models were not trained for similarity?
4 Correlation study on AmpC with public ROCS scores (200k molecules sampled across ROCS similarity bins): GeoDiff 2D PED aligns strongly with ROCS color similarity (Pearson r ≈ -0.60), GeoDiff 3D PED aligns with ROCS shape similarity (r ≈ -0.60), and concatenated GeoDiff (2D 3D) best matches ROCS combined similarity (r ≈ -0.67). MoLFormer PED also aligns well with ROCS combined similarity (r ≈ -0.63), with stronger correlation to color than shape, consistent with its SMILES/2D nature.
5 Virtual screening evaluation on LIT-PCBA (15 targets) uses EF1% and a best-pooled strategy across multiple reference ligands (min distance to any known ligand). Across targets, PED variants are competitive and often strong even on targets considered unfavorable for shape-based screening.
6 On LIT-PCBA with cosine distance, MoLFormer PED achieves the highest overall average mean EF1% (4.53 ± 2.79) and best-pooled EF1% (6.15), outperforming traditional baselines reported in the benchmark such as ECFP4 similarity (avg mean 3.94 ± 2.43; best-pooled 4.83) and several 3D similarity settings.
7 Using Euclidean distance yields similar conclusions: MoLFormer has the highest average mean EF1% (4.31 ± 3.11), while GeoDiff 3D Euclidean PED gives the best average best-pooled EF1% (5.47), suggesting complementary strengths depending on whether one optimizes typical or best-case retrieval across references.
8 PED is also used as a reward for RL-guided molecular generation, replacing expensive 3D alignment scoring. Two generators are tested: REINVENT (SMILES RL) and SynFormer (synthesizable pathway-based generation). PED distances are converted to bounded rewards via a reverse-sigmoid transformation; Euclidean PED is emphasized for reward shaping due to its dynamic range.
9 In a BTK inhibitor case study (reference: BMS-986195), PED rewards converge faster than 3D scoring and reduce compute overhead by avoiding conformer generation/alignment. Reported speedups in REINVENT sampling: ~1.5× (GeoDiff PED) and ~3.3× (MoLFormer PED) vs a ROSHAMBO2 3D-similarity reward; SynFormer sees ~2× faster sampling with PED-based rewards.
10 Quality trade-offs are analyzed via scaffold diversity, drug-likeness, and predicted binding potency (Boltz-2 pIC50). High-scoring scaffold diversity depends on both the embedding mode and generator: REINVENT shows high top-5k scaffold ratio with GeoDiff 3D (35.16%), while SynFormer shows highest with GeoDiff 2D (46.36%). For predicted potency on scaffold-balanced sets, REINVENT performs best with MoLFormer reward (pIC50 10.27 ± 1.34; large effect vs ROSHAMBO2), while SynFormer performs best with GeoDiff 2D reward (pIC50 8.81 ± 0.87). This suggests PED can be a strong similarity-based objective, but the optimal embedding choice can be framework-dependent.
📜Paper: arxiv.org/abs/2604.24474
#ComputationalChemistry #Cheminformatics #DrugDiscovery #VirtualScreening #MolecularGeneration #ReinforcementLearning #FoundationModels #MolecularRepresentationLearning #AIforScience #Bioinformatics
3
19
1,425
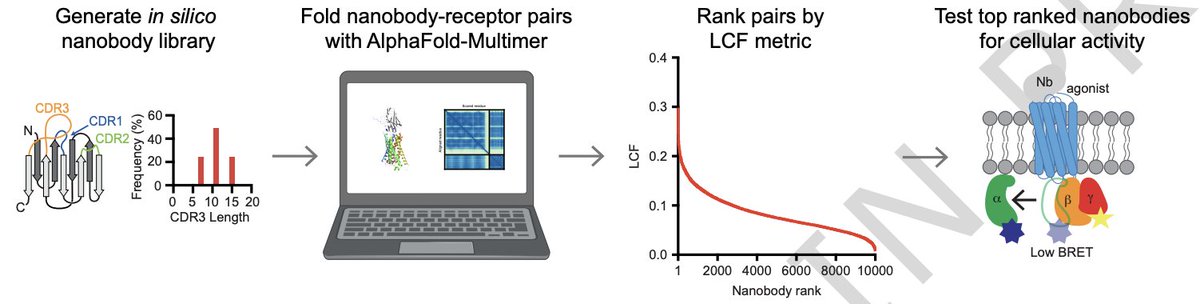
In silico discovery of nanobody binders to a G-protein coupled receptor using AlphaFold-Multimer @NatureComms
1. The study demonstrates a prospective, fully in silico nanobody discovery workflow that finds real GPCR binders: AlphaFold-Multimer (AF-M) screening of 10,000 synthetic VHH sequences yielded nanobody antagonists of the itch/inflammation GPCR target MRGPRX2, validated by cell binding and functional assays.
2. A key insight is that AF-M can separate binding vs non-binding nanobody–GPCR pairs using confidence/readout patterns from predicted complexes, despite the lack of antibody–antigen co-evolution. The authors benchmarked on curated post-training-cutoff structures/binding pairs and constructed matched negative controls by permuting nanobody–antigen pairings.
3. They identify AF-M outputs that are most informative for GPCRs (AUROC > 0.65 for several), including pTM and interface-focused metrics (interface PAE, interface pLDDT), plus pDockQ. They combine six scaled metrics into a single ranking score (LCF) intended to be more robust than any single metric.
4. For GPCRs, the approach is especially useful in the regime that matters for screening: the very top-ranked predictions. Precision among the top 5% (AUC5%) is extremely high (0.93–1.0 across top metrics/LCF), suggesting AF-M ranking can strongly enrich for true GPCR binders when only a small number of hits are tested experimentally.
5. The same AF-M strategy does not generalize well (yet) to soluble proteins or non-GPCR membrane proteins in their benchmarks: AUROCs hover near chance and top-5% precision is low (≤0.22). The authors attribute the GPCR advantage to the growing number of GPCR–nanobody structures in the PDB and more stereotyped binding modes on GPCR extracellular surfaces.
6. Prospective screen details: they generate 10,000 “naive” nanobody sequences matching a published yeast-display library design (CDR3 lengths 7/11/15; position-specific amino-acid distributions; largely excluding Cys/Met). AF-M predictions were run without templates to reduce bias, and sequences with developability liabilities (e.g., CDR glycosylation motifs, predicted polyreactivity) were filtered out.
7. From 10,000 designs, 179 sequences exceeded a threshold defined by the best negative-control LCF in the GPCR benchmark set; 177/179 were predicted to bind extracellularly (consistent with desired competition against endogenous ligands). Ten candidates spanning the ranking distribution were expressed as Fc fusions for testing.
8. Experimental validation: three top-ranked nanobodies (Sim8619 rank 1, Sim9877 rank 5, Sim4784 rank 7) bind MRGPRX2 on ROSA mast cells and on MRGPRX2-transfected HEK293T cells with nanomolar affinities. Reported Kd values include ~20–200 nM depending on clone and cell context; Sim8619 and Sim9877 show high specificity vs other peptide-binding GPCRs (MC4R, CXCR3), while Sim4784 shows some MC4R off-target binding.
9. Functional validation: the three binders are not agonists (no mast-cell degranulation on their own), but act as antagonists by suppressing compound 48/80-induced degranulation. AF-M models place them in the orthosteric pocket; targeted mutagenesis on nanobody (e.g., Sim8619 R102A) and receptor (E164/D184) supports predicted salt-bridge interactions for specific clones, and competition assays show reduced Gi signaling Emax and right-shifted EC50 for orthosteric agonists (48/80 and substance P).
💻Code: github.com/kruselab/MRGPRX2-…
📜Paper: doi.org/10.1038/s41467-026-7…
#ComputationalBiology #ProteinStructure #AlphaFold #Nanobody #AntibodyEngineering #GPCR #VirtualScreening #DrugDiscovery #Immunology #MachineLearning
1
11
97
14,389
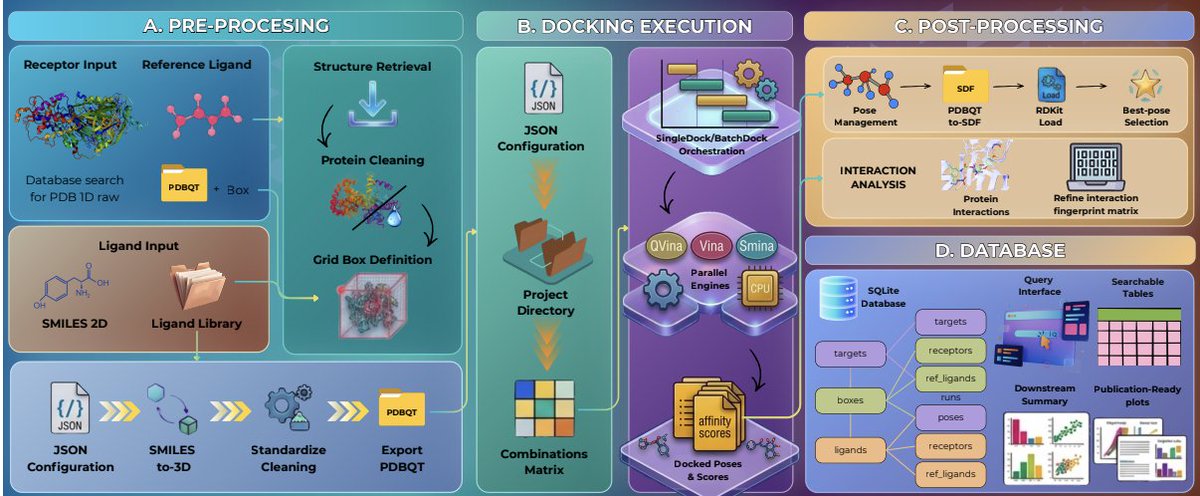
ProDock: From Multi-Target Consensus Docking into Database-Backed Storage
1. ProDock is an open-source Python toolkit that focuses on the workflow failures that often break real docking studies (preparation, organization, parsing, provenance), rather than proposing a new scoring function.
2. The core idea is to treat docking as an explicit many-to-many “campaign”: multiple receptors x multiple ligands x multiple docking backends, all captured in a machine-readable JSON plan that preserves parameters (e.g., seed, exhaustiveness, CPU, pose count).
3. ProDock connects four stages into one consistent, project-local pipeline: (i) receptor/ligand preprocessing, (ii) provenance-aware execution, (iii) postprocessing (poses, scores, interaction fingerprints), and (iv) SQLite-backed storage for later querying and comparison.
4. Receptor preprocessing supports PDB IDs or local files, with reproducible structure retrieval and curation (chain selection, optional cofactor retention, reference ligand extraction). Cleaning and fixing rely on PDBFixer, with optional minimization via OpenMM/Open Babel, and export to docking-ready PDBQT via Open Babel or Meeko.
5. Ligand preparation is separated from docking to enable reuse across targets: it accepts SMILES/tabular inputs or prepared directories, uses RDKit to generate 3D, add hydrogens, embed/optimize conformers, and exports engine-ready formats.
6. Search-space definition is integrated as a first-class preprocessing step via GridBox. Besides manual boxes, ProDock can auto-generate boxes from reference ligands using multiple algorithms (pad/advanced/percentile/PCA-based AABB/centroid-fixed), aiming to make “autoboxing” reproducible and auditable.
7. Execution is organized through SingleDock (single jobs) and BatchDock (screening-scale orchestration), with a common interface across engines (smina, vina, qvina2, qvina-w). Backend-specific details are normalized into standardized task definitions while still retaining engine parameters.
8. Postprocessing is treated as analysis, not an afterthought: PoseCrawler standardizes heterogeneous outputs (poses, ranks, engines, scores) into tables; InteractionProfiler integrates ProLIF to compute residue-level protein–ligand interaction fingerprints for interpretation beyond raw scores.
9. A key deliverable is database-backed docking: results can be inserted into a project-local SQLite DB (tables for receptors/ligands/engines/poses/scores/interactions), enabling fast queries by target, ligand, engine, rank, thresholds, or interaction patterns—without repeatedly reparsing raw output files.
10. Case study: an EGFR campaign across five crystal structures (2ITY, 1M17, 4G5J, 4I23, 4ZAU) starting from only PDB IDs ligand SMILES, with reference-ligand-based boxes and four engines (smina/vina/qvina/qvinaw), demonstrating multi-target/multi-engine orchestration and structured result collection.
💻Code: github.com/Medicine-Artifici…
📜Paper: arxiv.org/abs/2604.21828
#Bioinformatics #CompChem #MolecularDocking #VirtualScreening #Reproducibility #WorkflowAutomation #SQLite #Python #Cheminformatics
1
2
12
1,172

Salvador now screening on Venezia Shorts Virtual Channel. View here: veneziashortsvirtual.ottchan… Sign up for free membership to view. #henryiancusick #indieshort #virtualscreening

1
4
7
116
Structure-guided molecular design with contrastive 3D protein–ligand learning
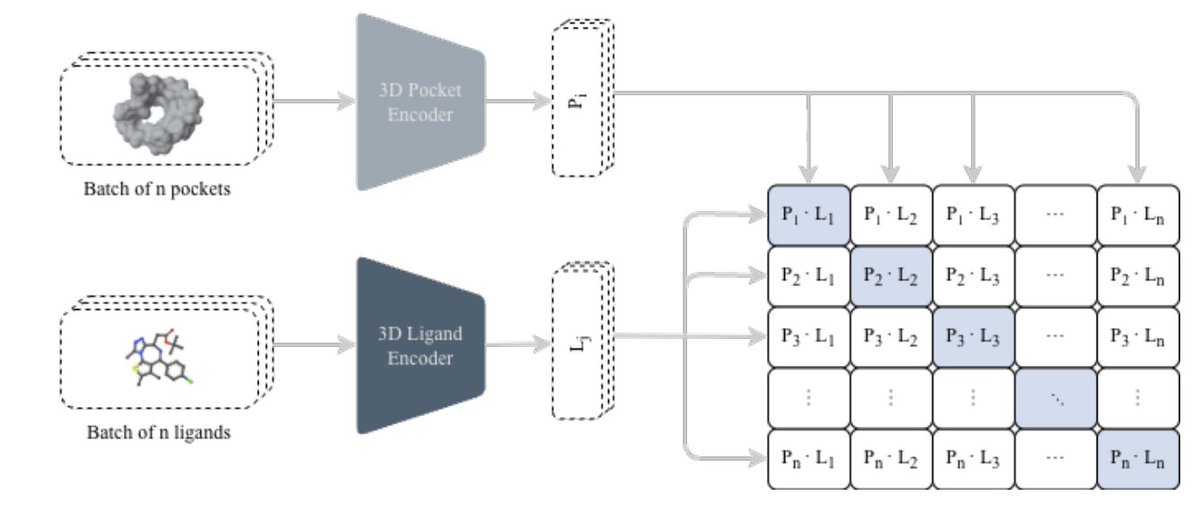
1 The paper proposes a unified pipeline that connects two usually separate steps in structure-based drug discovery: (i) fast 3D protein–ligand compatibility retrieval via contrastive learning, and (ii) de novo molecule generation via an autoregressive chemical language model, while explicitly steering outputs toward purchasable/synthetically accessible commercial spaces.
2 At the core is CLIPP-SET: an SE(3)-equivariant Transformer encoder that maps 3D pockets and 3D ligands into a shared embedding space, enabling cosine-similarity screening without docking ultra-large libraries and without target-specific fine-tuning (zero-shot).
3 A key technical detail is how they handle “false negatives” in contrastive learning caused by pocket collisions (many ligands bind the same pocket): they use Collision-Free InfoNCE (CF-InfoNCE), selecting (for pocket→ligand) the strongest-affinity ligand among those sharing the same pocket as the positive, rather than treating all off-diagonal pairs as negatives.
4 On LIT-PCBA zero-shot virtual screening (15 targets), CLIPP-SET pocket-based screening shows particularly strong early enrichment: best BEDROC, EF(0.5%), and EF(1%) among compared baselines, indicating it prioritizes actives near the top of the ranked list even if broader AUROC/EF(5%) can favor some docking-based methods.
5 The work then tests “realistic scale” retrieval on Enamine REAL (5.9B compounds): pocket-only queries (no reference ligand) retrieve top-100 candidates per target with higher predicted pIC50 on 13/15 targets and much higher internal diversity than 2D Morgan fingerprint search, suggesting the embedding captures binding-relevant geometry rather than just 2D similarity.
6 Because ligands and pockets share the same embedding space, the same system supports ligand-based search for scaffold hopping: given a known active, it retrieves molecules with high 3D shape/pharmacophore similarity but low 2D similarity (lower Tanimoto than Morgan FP), aligning with the goal of finding chemically novel scaffolds that preserve 3D binding features.
7 For generation, they introduce an MCLM (multimodal chemical language model): a Llama2-style autoregressive SMILES decoder conditioned by prepending a learned structural embedding token (from the frozen contrastive encoder) plus a learned dataset token [ds] that controls which chemical space distribution to emulate.
8 The dataset token is the mechanism for “commercial space steering”: trained on a 287M conformer–SMILES corpus labeled by source (PubChem, Enamine Diversity, Mcule, ChEMBL, GEOM-drugs), the model can be prompted with an “Enamine” token at inference to bias outputs toward Enamine-like chemistry; an ablation shows the token increases nearest-neighbor similarity to Enamine REAL and raises the fraction of exact catalog matches.
9 In structure-conditioned de novo design on LIT-PCBA (100 molecules/target), ligand-conditioned generation achieves the highest predicted affinity metrics (affinity probability and predicted pIC50) among evaluated methods, while pocket-conditioned generation (despite the decoder being trained only with ligand embeddings) remains competitive without reference ligands and yields the highest diversity, highlighting practical utility when only target structure is available.
📜Paper: arxiv.org/abs/2604.19562
#ComputationalBiology #Cheminformatics #DrugDiscovery #StructureBasedDrugDesign #GenerativeAI #MachineLearning #ProteinLigand #EquivariantNetworks #ContrastiveLearning #VirtualScreening
1
6
25
7,268
Structure-guided molecular design with contrastive 3D protein–ligand learning
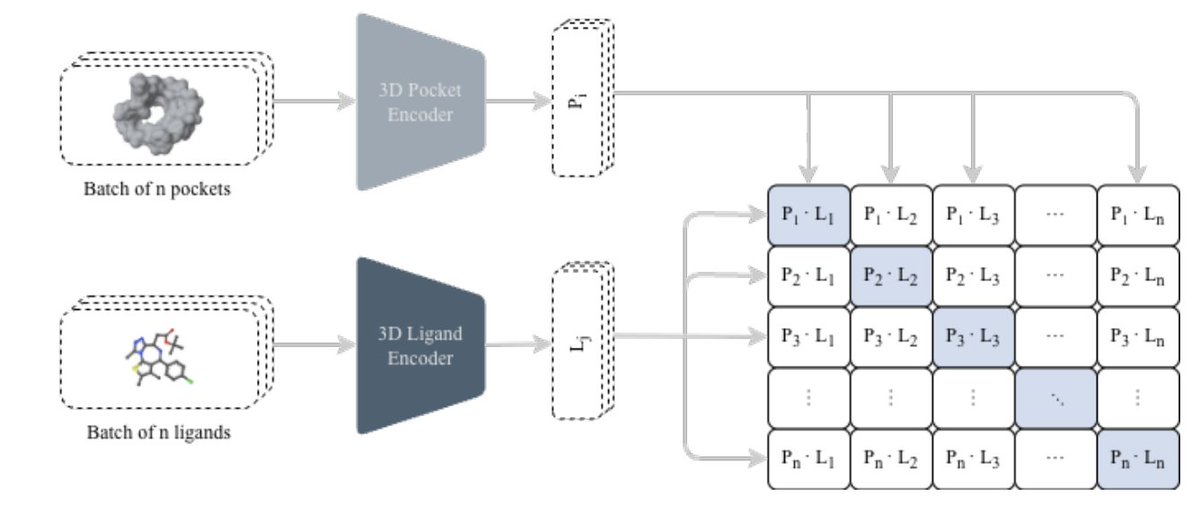
1 The paper proposes a unified pipeline that connects two usually separate steps in structure-based drug discovery: (i) fast 3D protein–ligand compatibility retrieval via contrastive learning, and (ii) de novo molecule generation via an autoregressive chemical language model, while explicitly steering outputs toward purchasable/synthetically accessible commercial spaces.
2 At the core is CLIPP-SET: an SE(3)-equivariant Transformer encoder that maps 3D pockets and 3D ligands into a shared embedding space, enabling cosine-similarity screening without docking ultra-large libraries and without target-specific fine-tuning (zero-shot).
3 A key technical detail is how they handle “false negatives” in contrastive learning caused by pocket collisions (many ligands bind the same pocket): they use Collision-Free InfoNCE (CF-InfoNCE), selecting (for pocket→ligand) the strongest-affinity ligand among those sharing the same pocket as the positive, rather than treating all off-diagonal pairs as negatives.
4 On LIT-PCBA zero-shot virtual screening (15 targets), CLIPP-SET pocket-based screening shows particularly strong early enrichment: best BEDROC, EF(0.5%), and EF(1%) among compared baselines, indicating it prioritizes actives near the top of the ranked list even if broader AUROC/EF(5%) can favor some docking-based methods.
5 The work then tests “realistic scale” retrieval on Enamine REAL (5.9B compounds): pocket-only queries (no reference ligand) retrieve top-100 candidates per target with higher predicted pIC50 on 13/15 targets and much higher internal diversity than 2D Morgan fingerprint search, suggesting the embedding captures binding-relevant geometry rather than just 2D similarity.
6 Because ligands and pockets share the same embedding space, the same system supports ligand-based search for scaffold hopping: given a known active, it retrieves molecules with high 3D shape/pharmacophore similarity but low 2D similarity (lower Tanimoto than Morgan FP), aligning with the goal of finding chemically novel scaffolds that preserve 3D binding features.
7 For generation, they introduce an MCLM (multimodal chemical language model): a Llama2-style autoregressive SMILES decoder conditioned by prepending a learned structural embedding token (from the frozen contrastive encoder) plus a learned dataset token [ds] that controls which chemical space distribution to emulate.
8 The dataset token is the mechanism for “commercial space steering”: trained on a 287M conformer–SMILES corpus labeled by source (PubChem, Enamine Diversity, Mcule, ChEMBL, GEOM-drugs), the model can be prompted with an “Enamine” token at inference to bias outputs toward Enamine-like chemistry; an ablation shows the token increases nearest-neighbor similarity to Enamine REAL and raises the fraction of exact catalog matches.
9 In structure-conditioned de novo design on LIT-PCBA (100 molecules/target), ligand-conditioned generation achieves the highest predicted affinity metrics (affinity probability and predicted pIC50) among evaluated methods, while pocket-conditioned generation (despite the decoder being trained only with ligand embeddings) remains competitive without reference ligands and yields the highest diversity, highlighting practical utility when only target structure is available.
📜Paper: arxiv.org/abs/2604.19562
#ComputationalBiology #Cheminformatics #DrugDiscovery #StructureBasedDrugDesign #GenerativeAI #MachineLearning #ProteinLigand #EquivariantNetworks #ContrastiveLearning #VirtualScreening
3
9
897

Apr 21
Here I thought I couldn’t go to any screenings this week…
@MsMiaMendez @StevenTrunce @DrEricRodgers @realDaveFeldman @wideeyetv @metcollective
#CholesterolCodeMovie #VirtualScreening #Meetup
Mar 30

Watch Party: The Cholesterol Code
We’re streaming together and discussing what it actually means for keto and metabolic health.
Watch live or stream on your own to support the film.
Bring questions. Stay for discussion.
meetup.com/mentalhealthandth…

1
10
535
Identification of Potential Multitarget Directed Ligands for Alzheimer’s Disease by Coupling Virtual Screening and Experimental Validation #VirtualScreening #MolecularDynamics
pubs.acs.org/doi/10.1021/acs…
@Paulina_valhor @CeciliaScorza
#JCIM Vol66 Issue5 #PharmaceuticalModeling
2
6
551

Apr 12
Accelerating Drug Discovery with HyperLab: An Easy-to-Use AI-Driven Platform
biorxiv.org/content/10.1101/…
Summary:
HyperLab (by HITS) is an AI-driven, web-based drug discovery platform that integrates the full structure-based workflow—from protein–ligand prediction and ultra-large virtual screening (up to trillions of compounds) to molecular design, SAR, and ADME/T prediction—into a single, user-friendly interface. Powered by physics-informed deep learning, it delivers competitive accuracy (approaching AlphaFold3) with significantly faster computation. Experimental validation shows real-world impact, identifying potent compounds (IC50 ~70–600 nM) within 24 hours and enabling rapid optimization. HyperLab lowers the barrier to AI-driven drug discovery by making advanced computational tools accessible to experimental researchers.
#AIDrugDiscovery #HyperLab #StructureBasedDrugDesign #SBDD #DrugDiscovery #ADMET #VirtualScreening #AIBio
1
5
45
2,642
Accelerating Drug Discovery with HyperLab: An Easy-to-Use AI-Driven Platform
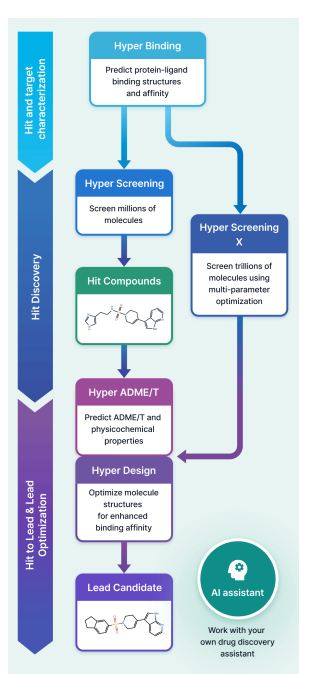
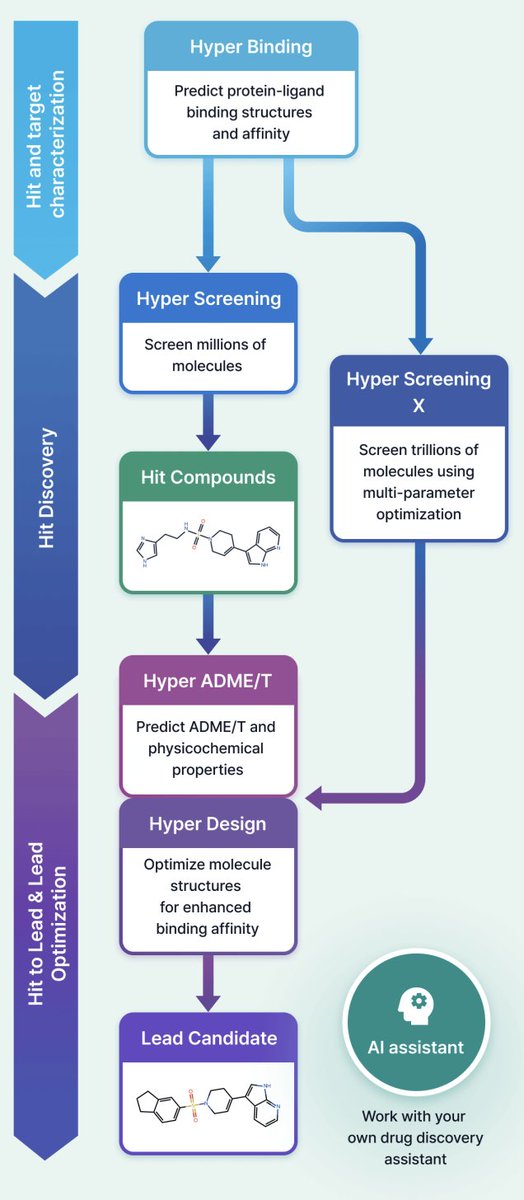
1 HyperLab (by HITS) is presented as a web-based, AI-driven SBDD platform aimed at making structure-based workflows usable by experimental drug discovery researchers without requiring AI/CADD expertise, emphasizing integrated UI/UX over fragmented toolchains.
2 The platform compresses early discovery into a single environment spanning: protein–ligand pose affinity prediction (Hyper Binding), covalent complex modeling (Covalent Hyper Binding), virtual screening from 1M to 11T compounds (Hyper Screening / Hyper Screening X), structure-based molecular optimization (Hyper Design), SAR analysis, and 19-endpoint ADME/T prediction (Hyper ADME/T), with an embedded AI assistant for workflow automation.
3 Hyper Binding’s key technical angle is physics-informed deep learning for protein–ligand interactions, supporting multiple protein inputs (PDB ID, uploaded PDB, AlphaFold structures via UniProt) and an end-to-end co-folding mode that predicts complex structures directly from protein sequence plus ligand, reducing dependence on curated receptor structures.
4 On PoseBuster v2 (PB-valid) pose prediction, Hyper Binding reports 77% accuracy when given binding-site information, compared with 58% for Vina and 13% for DiffDock; it approaches AlphaFold3 (84%) and is comparable to Boltz2 (78). The paper also highlights throughput: ~3 minutes per complex (via cloud) vs ~15 minutes for AlphaFold3 on an RTX 3060.
5 For binding affinity prediction on two FEP-style benchmarks (focused on subtle potency differences among close analogs), Hyper Binding reports Pearson r = 0.70 and 0.53, outperforming evaluated deep learning scorers (Luminet, GenScore) and physics-based docking (Glide SP, Vina) on both datasets.
6 Covalent drug discovery is treated as a first-class workflow: covalent pose prediction is benchmarked on a curated covalent set (from PDBBind/PDB). Covalent Hyper Binding (cofolding) reports 88.7% pose accuracy vs 48.4% (COV SMINA) and 46.8% (GNINA); the docking mode reports 61.3%. Screening enrichment (EF@10%) is reported as 6.56 (Mpro) and 9.97 (KRAS), exceeding baselines under the described setup.
7 Hyper Screening targets rapid hit finding by running Hyper Binding across curated libraries and returning top-ranked candidates (top 500). Built-in libraries include: Diverse (1,000,000), Fragment (500,000; rule-of-three-like), Kinase-focused (65,000), Natural product-like fragments (4,200), and FDA-approved (1,100), plus support for user-registered libraries.
8 Hyper Screening X expands to an 11-trillion-molecule virtual space using generative exploration with GFlowNet-based models, optimizing binding score plus properties (e.g., MW, TPSA, LogP). The workflow is described as: set target property constraints, train (~48h), then generate molecules (e.g., 100 molecules in ~30 min), with synthetic route output and optional synthesis request via a partner service.
9 Hyper Design provides structure-based optimization starting from a scaffold or an X-ray-bound ligand, enabling user-specified modification sites and fragment growth/replacement with synthesizability constraints; outputs include 3D structures and iterative “design trees.” The paper positions use cases as fragment-to-lead growth and generating patent-distinct analogs while preserving key interactions.
10 The internal validation study emphasizes “no post-analysis/visual inspection” selection: a 24-hour Hyper Screening run led to 52 compounds tested, yielding 5 hits with IC50 70–600 nM (~9% hit rate). Hyper Design then produced derivatives; 5 were synthesized and 3 showed >75% inhibition at 1 µM with IC50 200–400 nM, including one compound comparable or better than a reference and with supporting pathway assay readouts.
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #ComputationalBiology #Cheminformatics #StructureBasedDrugDesign #VirtualScreening #CovalentInhibitors #ADMET #GenerativeAI #ProteinLigandDocking #BioRxiv
3
16
1,460
Rapid Parallel Virtual Screening Aids the Discovery of Novel P-Glycoprotein Inhibitors #VirtualScreening #DrugDiscovery
pubs.acs.org/doi/10.1021/acs…
@Konrad_Pakula @CarstenUhd
#JCIM Vol66 Issue5 #compchem
3
19
930
UniDock-Pro: A Unified GPU-Accelerated Platform for High-Throughput Structure-Based, Ligand-Based, and Synergistic Hybrid Virtual Screening #VirtualScreening
pubs.acs.org/doi/10.1021/acs…
#JCIM Vol66 Issue5 #compchem
3
21
903