
Useful package for enumerating reaction prodcuts from building block dataset #cheminformatics #rdkit #memo iwatobipen.wordpress.com/202… @iwatobipenより
1
10
538

DeepRHP: A Hybrid Variational Autoencoder for Designing Random Heteropolymers as Protein Mimics
1. The paper introduces DeepRHP, a semi-supervised hybrid VAE that learns latent representations of random heteropolymer (RHP) sequence ensembles while explicitly constraining the latent space to reflect function-related chemical features, aiming to make RHP design more data-driven than empirical screening.
2. Key architectural idea: a classical sequence VAE is paired with a parallel feature-based VAE that reconstructs a deterministic chemical feature y derived from the same sequence x; both branches share the same latent variable z, encouraging z to encode both sequence-pattern statistics and chemically meaningful structure.
3. The training objective modifies the standard VAE ELBO by combining two reconstruction terms: (a) discrete sequence reconstruction (cross-entropy over monomer tokens) and (b) feature reconstruction (MSE on y), weighted by a tunable α, while keeping the KL regularization on q(z|x) vs p(z).
4. The “feature” used for semi-supervision is the sliding-window average hydrophilic–lipophilic balance (HLB), motivated by prior evidence that local hydrophobicity/solubility patterning is strongly tied to RHP behavior in protein stabilization and transport applications.
5. Data pipeline: the study simulates 10,000 RHP sequences per monomer composition using Compositional Drift (copolymer models Monte Carlo), focusing on a 4-methacrylate monomer set (MMA, EHMA, OEGMA, SPMA) spanning hydrophobic, very hydrophobic, hydrophilic, and charged chemistries.
6. To connect synthetic polymers to biology, ~30k membrane and ~30k globular protein sequences (UniProt, 50% identity threshold) are reduced into a 4-letter “monomer-equivalent” alphabet based on residue hydrophobicity/charge, enabling joint embedding and similarity analysis between proteins and RHP ensembles.
7. Design insight 1 (alphabet size): by comparing 2-monomer vs 4-monomer RHP libraries in the learned latent space (visualized via PCA of latent factors), the paper argues that 2-monomer sequence space is too broad relative to protein-like regions, whereas 4-monomer libraries yield more localized, protein-overlapping distributions—supporting why four monomers can be “enough” for protein-mimic behavior.
8. Design insight 2 (composition): within a fixed 70% hydrophobic / 30% hydrophilic constraint, varying the MMA:EHMA ratio produces distinct RHP ensembles; DeepRHP’s latent-space overlap with Aquaporin Z (AqpZ) projections highlights specific compositions (notably matching the published optimal formulation) as most similar to the target membrane protein.
9. Practical takeaway: DeepRHP reframes RHP design as an ensemble-level representation learning problem—enabling composition suggestion by latent-space similarity to target proteins—without requiring exact polymer sequences, 3D structures, or multiple sequence alignment, and with a plug-in pathway to incorporate other chemical features beyond HLB.
10. The authors report ablations indicating the hybrid (feature-guided) architecture outperforms a classical VAE alone for producing useful latent structure, while noting that current evaluation is largely qualitative and motivating future quantitative metrics and downstream tasks (e.g., membrane protein subclass discrimination, RHP–protein similarity scoring).
📜Paper: arxiv.org/abs/2606.11651
#ComputationalBiology #MachineLearning #DeepLearning #VAE #GenerativeModels #PolymerScience #MaterialsInformatics #ProteinEngineering #MembraneProteins #Cheminformatics

1
13
947
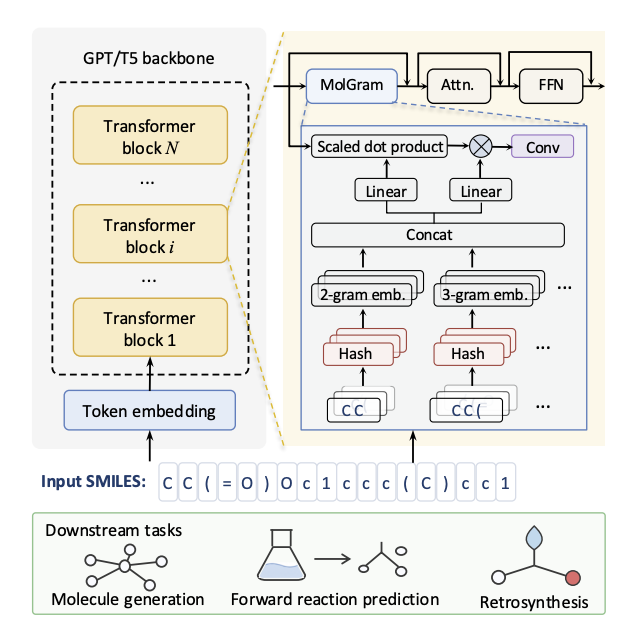
Augmenting Molecular Language Models with Local n-gram Memory
1. The paper identifies a “locality gap” in SMILES Transformers: character-level tokenization splits chemically meaningful motifs (e.g., C(=O), aromatic rings) across many tokens, so model capacity gets spent relearning local syntax instead of global structure and reaction logic.
2. MolGram is introduced as a model-side fix that keeps standard character tokenization unchanged, but augments the Transformer with a conditional local n-gram memory to explicitly store and retrieve recurring SMILES patterns.
3. Core mechanism: local n-grams are mapped to learned embeddings using scalable hash-based lookup (O(1) access, no explicit n-gram dictionary), with multi-head hashing and prime-sized tables to reduce collisions and capture diverse pattern aspects.
4. Contextual control: retrieved n-gram embeddings are static, so MolGram adds a substructure-aware gate conditioned on the backbone hidden state. It uses RMSNorm scaled dot product to score relevance, then a sign-preserving sqrt sigmoid to stabilize gating and avoid vanishing gradients.
5. Local composition: after gating, a lightweight depthwise convolution mixes neighboring retrieved signals so adjacent motifs can combine into higher-order chemistry (e.g., carbonyl hydroxyl patterns supporting carboxylic-acid-like contexts), and the result is added residually back to the hidden state.
6. Architecture-specific integration: for decoder-only MolGPT, MolGram is inserted into selected decoder layers with causal convolution to preserve autoregressive generation; for encoder-decoder T5Chem, it is inserted into early encoder layers so cross-attention benefits from substructure-enriched source representations.
7. Unconditional molecule generation (MOSES, GuacaMol): MolGram improves validity/uniqueness/novelty and distributional quality (FCD-related metrics). A smaller MolGram (GPT, 27.2M) can outperform a much larger baseline MolGPT (85.4M) on many generation metrics, suggesting efficiency beyond simple scaling.
8. Forward reaction prediction (USPTO-MIT, separated and mixed inputs): MolGram improves top-k accuracy across both GPT and T5 backbones. Notably, MolGram (T5, 15.0M) exceeds T5Chem (44.1M), and MolGram (GPT, 26.1M) matches/exceeds MolGPT (85.4M), reinforcing the “local memory as inductive bias” argument.
9. Single-step retrosynthesis (USPTO-50k): MolGram improves top-1 accuracy over both MolGPT and T5Chem, and smaller MolGram variants beat larger baselines (e.g., MolGram GPT 6.7M > MolGPT 25.5M; MolGram T5 15.7M > T5Chem 44.1M), consistent with local priors being especially useful when data is limited.
10. Interpretability signal: gate-activation visualizations show stronger MolGram contributions at chemically meaningful positions (aromatic interiors, functional groups like nitro and amide, halides, heteroatoms) and weaker activations on less informative tokens (brackets, some ring-closure endpoints, isolated aliphatic carbons).
📜Paper: arxiv.org/abs/2606.12113
#ComputationalChemistry #Cheminformatics #SMILES #Transformers #MolecularGeneration #ReactionPrediction #Retrosynthesis #NLP #DeepLearning
12
812

Jun 12
The chemistry filters are through the roof. Any cheminformatics query, any query involving SMILES or molecule, and that's it, we're back with Opus 4.8.
93

Jun 12
🥳Check out our latest paper "Assigning the stereochemistry of natural products by machine learning" by Markus Orsi and @jrjrjlr in Journal of Cheminformatics! Read the full story: link.springer.com/article/10…
@DCBPunibern
@reymondgroup
@unibern

2
11
712

A most excellent molecule (and chemist) with which to connclude the week #SynChem #BB #MedChem #DrugDesign #DrugDiscovery #cheminformatics
Jun 12

Please welcome Ivan Mazurenko - the chemist behind the molecule of this week. Congratulations!👏
enaminestore.com/catalog/EN3…
Discover all the molecules of the week here: enamine.net/molecule-of-the-…
#Enamine #molecule #chemistry #science #drugdiscovery

44

Jun 12
#descriptoroftheday #moleculardescriptors #qsar #cheminformatics #alvatips
Kier alpha-modified shape indices have been described in Kier, L. B. (1986) doi.org/10.1002/qsar.1986005… and Kier, L. B. (1986) doi.org/10.1002/qsar.1986005…
Check #alvaDesc at: alvascience.com/alvadesc/

15
Yuta Aoki, D.Sc. retweeted

Jun 10
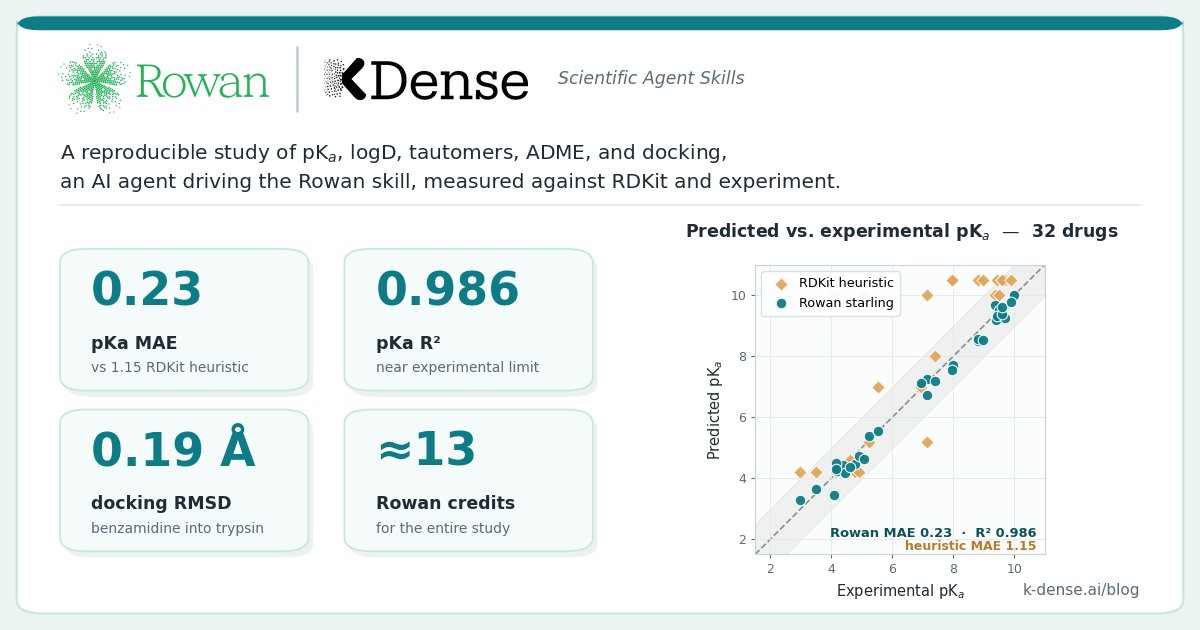
Every computational chemist has RDKit open in a terminal. Free, fast, local, genuinely excellent at cheminformatics. But ask it for a pKa and it has no answer. Ask for logD at pH 7.4 and it hands you a pH-blind logP. So we benchmarked the Rowan agent skill against experiment.
4
6
25
6,079

Ronald Clarke retweeted

19 Jan 2022
Working with #cheminformatics, #computational #metabolomics, and/or #retrosynthesis, and #Python workflows?! Consider using #PIKACHU to #visualise #molecular #structures and #reactions! 😎 Congratulations to Barbara, Sophie, and @marnixmedema! 👏
researchsquare.com/article/r…
1
12
25
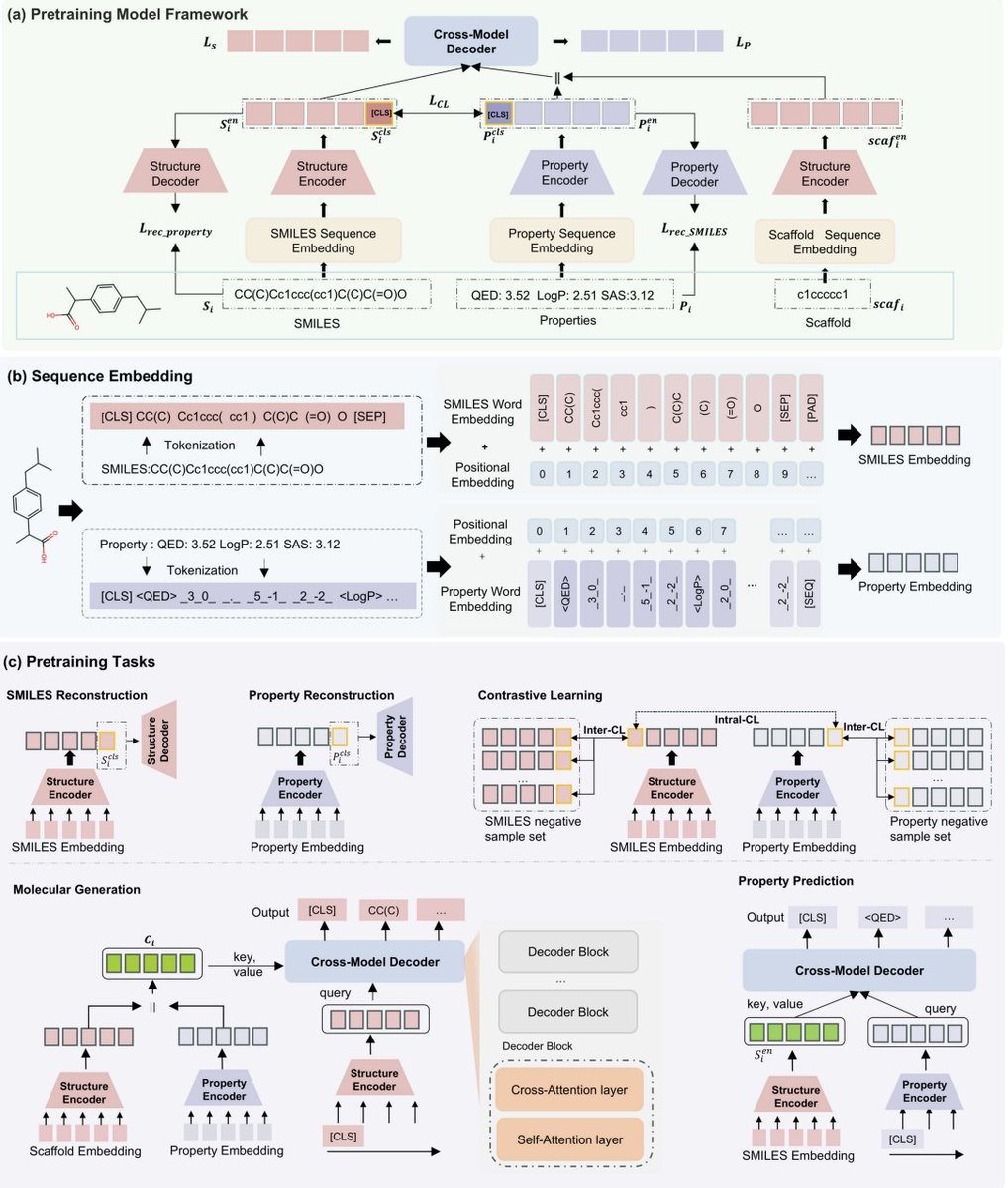
Generative pretraining for drug molecule design with bidirectional structure-property optimization
1. The paper presents BiSP-GP, a single pretrained framework that supports both controllable molecule generation (properties and/or scaffolds as conditions) and SMILES-to-property prediction, using one unified autoregressive sequence modeling setup rather than separate task-specific models.
2. A key idea is to turn continuous properties into “language”: QED, LogP, and SAS are serialized into semantic token sequences (property identifier, sign, digits, decimal point, and digit position tokens). This keeps numerical precision while letting properties be modeled in the same token space as SMILES, avoiding the usual “properties as plain numeric constraints” design.
3. Architecture: dual Transformer encoders (structure encoder for SMILES/scaffolds; property encoder for property-token sequences) plus a cross-modal decoder with cross-attention. The decoder enables bidirectional mapping: (a) generate SMILES conditioned on properties/scaffolds, and (b) generate property tokens conditioned on SMILES.
4. Pretraining uses five self-supervised objectives: SMILES reconstruction, property reconstruction, cross-modal intra-modal contrastive learning, conditional SMILES generation, and SMILES-conditioned property generation. The contrastive part includes a soft-label strategy (via momentum encoder) to reduce false negatives among structurally similar molecules with similar properties.
5. Robustness mechanism: stochastic masking of conditions. With 50% probability, an entire property’s tokens are replaced by [UNK], exposing the model to missing/incomplete property settings and enabling flexible inference-time control (choose which properties to constrain by providing tokens; leave others as [UNK]).
6. Unconditional generation (1,000 samples) is compared to CharRNN, LatentGAN, MolGPT, SPMM, and GP-MoLFormer. BiSP-GP reports the best composite V*U*N*I score (0.804) with strong validity (0.986), near-perfect uniqueness (0.999), high novelty (0.926), and high internal diversity (0.882), aiming for a better novelty–diversity balance than several baselines.
7. Single-property conditional generation (targets across QED, LogP, SAS) is evaluated with mean absolute deviation (MAD) for control accuracy plus Moses quality metrics. BiSP-GP shows the lowest MAD across all three properties versus CMGN, Scaffold-GGM, and SPMM, while maintaining strong uniqueness and internal diversity under constraints.
8. Multi-property control is tested for QED-LogP, QED-SAS, LogP-SAS, and QED-LogP-SAS conditions. The model maintains validity/uniqueness/novelty > 0.9 across scenarios and produces property distributions clustered around targets, while leaving unconstrained properties broadly distributed—useful for realistic multi-objective optimization.
9. Scaffold-conditioned and scaffold property generation: on 100 unseen scaffolds, BiSP-GP keeps scaffold similarity ratio (Sim_ratio) > 0.8 while generating novel variants; similarity analyses suggest novelty comes from both out-of-distribution scaffolds and side-chain diversification. Joint scaffold multi-property constraints still preserve scaffold structure with property values concentrated near targets.
10. Practical case study: PAK1 inhibitor optimization. With a fixed scaffold and a reduced LogP target (from 4.70 down toward 2.50 while holding QED and SAS), generated candidates show improved docking scores on PAK1 (PDB: 4EQC) on average (~0.35 kcal/mol better than the reference) and introduce additional polar interactions while retaining a key H-bond with GLU-315.
11. Property prediction as sequence generation: on 1,000 unseen molecules, BiSP-GP generates grammatically valid property strings and achieves very high agreement with RDKit-computed values (R²: LogP 0.999, QED 0.997, SAS 0.987). It remains reliable on randomized SMILES, suggesting learned structure–property relationships are not brittle to SMILES syntax variation.
12. Transfer learning: using the pretrained structure encoder as a frozen feature extractor plus a lightweight head, BiSP-GP performs strongly on MoleculeNet tasks plus Malaria and CEP, with statistically supported gains over several baselines on many regression/classification datasets; y-scrambling checks indicate performance is not driven by label artifacts.
13. Ablations indicate both innovations matter: replacing property serialization with numeric embeddings degrades conditional control (notably LogP MAD) and lowers property-prediction R²; removing contrastive learning broadly reduces generation quality, controllability, and prediction accuracy—supporting the role of cross-modal alignment.
💻Code: github.com/xmubiocode/BiSP-G… (Zenodo: zenodo.org/records/20115955)
📜Paper: doi.org/10.1038/s42004-026-0…
#ComputationalChemistry #Cheminformatics #MolecularGeneration #DrugDiscovery #Transformers #FoundationModels #GenerativeAI #PropertyPrediction #ScaffoldHopping #RepresentationLearning
2
23
1,588
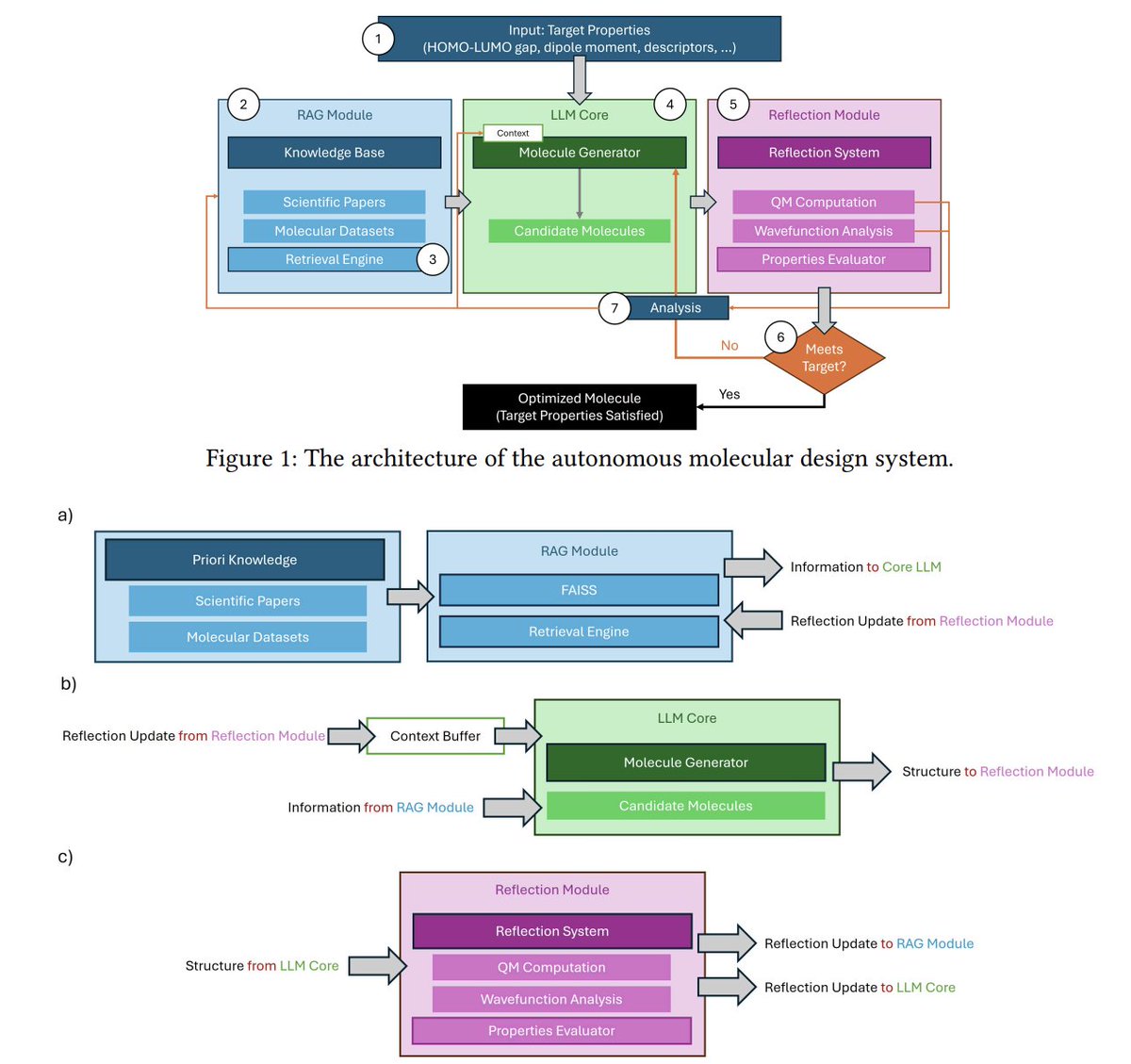
Closing the Prior-Posterior Loop: Self-Reflective Molecular Design with Analysis-Driven LLM Iteration
1. The paper proposes an alternative to “Generate → Score → Regenerate” in LLM molecular design: “Generate → Analyze → Reflect → Refine”, where the model is fed mechanism-level quantum-chemistry evidence (e.g., orbital energies, charges, electron density) instead of a single scalar score.
2. Core claim: providing full physicochemical rationale from first-principles calculations can shift an LLM’s behavior from stochastic sampling toward more causal, structure-property reasoning—because the model learns not only that a candidate misses the target, but why.
3. System architecture has three coupled parts: (i) a retrieval-augmented generation (RAG) module for prior knowledge, (ii) an LLM core that proposes candidates, and (iii) a reflection module that runs quantum calculations and converts raw outputs into actionable design edits.
4. The RAG database is built from QM9 (about 130k small organic molecules, <9 heavy atoms) using a FAISS vector index; retrieval is conditioned on the requested target property (e.g., HOMO-LUMO gap).
5. The reflection module explicitly avoids treating computation as a black-box scorer. It preserves rich outputs such as HOMO/LUMO energies, Mulliken charges, total electronic energies, dipole moments, and (conceptually) wavefunction/electron-density information.
6. For efficiency, evaluation is staged: GFN2-xTB is used for geometry optimization and fast pre-screening, then pySCF performs higher-accuracy DFT on top candidates (default batch: x=20 candidates screened, y=5 sent to DFT).
7. The self-reflection procedure is described as a 3-step pipeline: (1) extract key parameters from DFT output, (2) perform causal reasoning linking structure to the target property, (3) plan concrete structural modifications for the next iteration; reflection insights are also written back into the RAG context.
8. On targeted HOMO-LUMO gap design across 5 targets (5.0, 4.0, 3.0, 2.0, 1.0 eV), SPR reflection (mechanism-level feedback) RAG is consistently the most stable configuration; for the 3.0 eV task it reports deviation down to 0.0003 eV, and for the 2.0 eV task it is the only configuration reaching 100% success rate (within the authors’ success definition).
9. The paper highlights a failure mode of scalar-only feedback: on the hardest 1.0 eV gap target, Scalar RAG fails (0/3 successes), while SPR RAG yields at least one close solution (0.0164 eV deviation), suggesting that “far from target” numbers alone may not provide an actionable gradient for difficult design regimes.
10. Additional findings: (i) convergence is not monotonic—extra iterations can cause “overthinking” and oscillations; (ii) batch reflection can outperform per-molecule reflection (BFS-like vs DFS-like exploration); (iii) the framework generalizes beyond gaps to dipole-moment targeting (example target 2.5 D, best deviation ~0.016 D), and appears robust across five LLM backbones (DeepSeek-V4Pro/Flash, MiniMax-M3, Qwen-3.7Max, GLM5.1).
📜Paper: arxiv.org/abs/2606.09520
#ComputationalChemistry #MolecularDesign #LLM #RAG #QuantumChemistry #DFT #InverseDesign #AIforScience #Cheminformatics
2
6
1,085
Jun 10
#descriptoroftheday #moleculardescriptors #qsar #cheminformatics #alvatips
Kier alpha-modified shape indices have been described in Kier, L. B. (1986) doi.org/10.1002/qsar.1986005… and Kier, L. B. (1986) doi.org/10.1002/qsar.1986005…
Check #alvaDesc at: alvascience.com/alvadesc/

20

Jun 10
Synthesis planning is where a lot of drug programs quietly die. You design a beautiful molecule, then spend months figuring out how to actually make it.
ReBolt (our synthetic chemistry platform) automates multi-step retrosynthesis using ML models trained on massive reaction datasets — template-based and template-free, transformers and GNNs, reinforced with Monte Carlo Tree Search.
You get ranked routes with scores, estimated yields, cost projections, and direct vendor links to Sigma-Aldrich and MolPort.
From SMILES to purchasable starting materials without leaving the platform.
#retrosynthesis #drugdiscovery #cheminformatics
3
118
Opportunity for computational chemist at Vertex (Oxford UK) ref: REQ-29266 | closing: 19-Jun-2026 #CompChem #cheminformatics #ML #AI #UKChemJobs #ChemJobs
vrtx.wd501.myworkdayjobs.com…
74

This is great to hear, I’ve been hoping for some way of identifying oneself as a legit scientist for bio research prone to tripping filters. (Bio)chemistry too! Small molecule cheminformatics is less prone than bioinformatics in my experience, but still can be difficult.
1
165