
the new txdr is kinda lax when it comes to that. remember that one game that was literally impossible to 100% cause of a programmig error preventing you from getting enough cash? or having to do rally stages in drift 2? pain indeed
1
8

Jun 10
They have more time... World was not such complicated and hurry like these days. I was pretty normal to have book, read about programmig language and hack own code. Today it is not acceptable to study, understand and implement something by hand.
133

Jun 9
Morning chat ,forgot to post yesterday's progress...so here this is
--Completed ch 9 and 10 , learnt about transistor switching and capacitors
--completed recursion portion from Grokking Algo
--started revising digital electronics
--read a blog about bare metal programmig


40

STOP putting ur focus everywhere but here.!
👇🏼👇🏼👇🏼👇🏼👇🏼👇🏼👇🏼👇🏼👇🏼
#StevenDKelley and
#OccupyTheGetty Mission
Is the answer!!!
We stop this merry go round of polished Hollywood bullshitters who add to your programmig when everyone focus on the #Getty
#MelGibson , #TimBallard #Cabaziel fake Jesus
a few examples of this matrix distractions, while children are being consumed by these satanists POS every single day on an industrial scale.
#MelGibson is a fraud.
@TimBallard the protagonist of The Sound of Bullshit is another fraud, six counts of sexual assault by six different women,
x.com/jimstewartson/status/1…
☝️
This what an #Intertel #NSA #operative asset looks like.
“Programming, or Ignorance?.”
June 8th 2023
The
Steven D Kelley Show
TruthCatRadio.com
Join the fight:
t.me/OfficialOccupyTheGettyP…

Which means that we shall be many to the enemy's few.
4
6
207

May 20
what do you mean "a lot" ? Do any other programmig language actually exist?
1
184

May 11
I honestly agree with you here, well if you were told to write a program with your favorite program that would probably be super fan to do.
money is what got me into the game but that doesn't mean i still don't like doing programmig.
2
340

May 11
My initial reaction was to bookmark this for later, but then I thought of all the other "this is the only markdown you need" or "god himself uses this claude.md" which I tried and saw no difference or found some of the "rules" annoying in practice. I always ended up with guiding claude my own way, which is to treat it like a super smart junior dev who was top of his class, read all the programmig books and can do incredible things but has zero work experience and can (and will) still make the stupidest mistakes here and there, so I need to check his work all the time because I'm his manager and in the end I will be responsible for the code. And it works, but the "programmers are obsolete" bullshit has never been more laughable in my view :).
So I didn't bookmark it and while not bookmarking it, I saw your comment which confirmed my instinct ;)
1
7
377

May 7
6. CS50's Introduction to Programming with Python.
Explore programming using Python for various applications like:
- Data science
- Web programmig
- General-purpose programming
🔗 hedx.sjv.io/qz0j9y
1
4
240


Apr 6
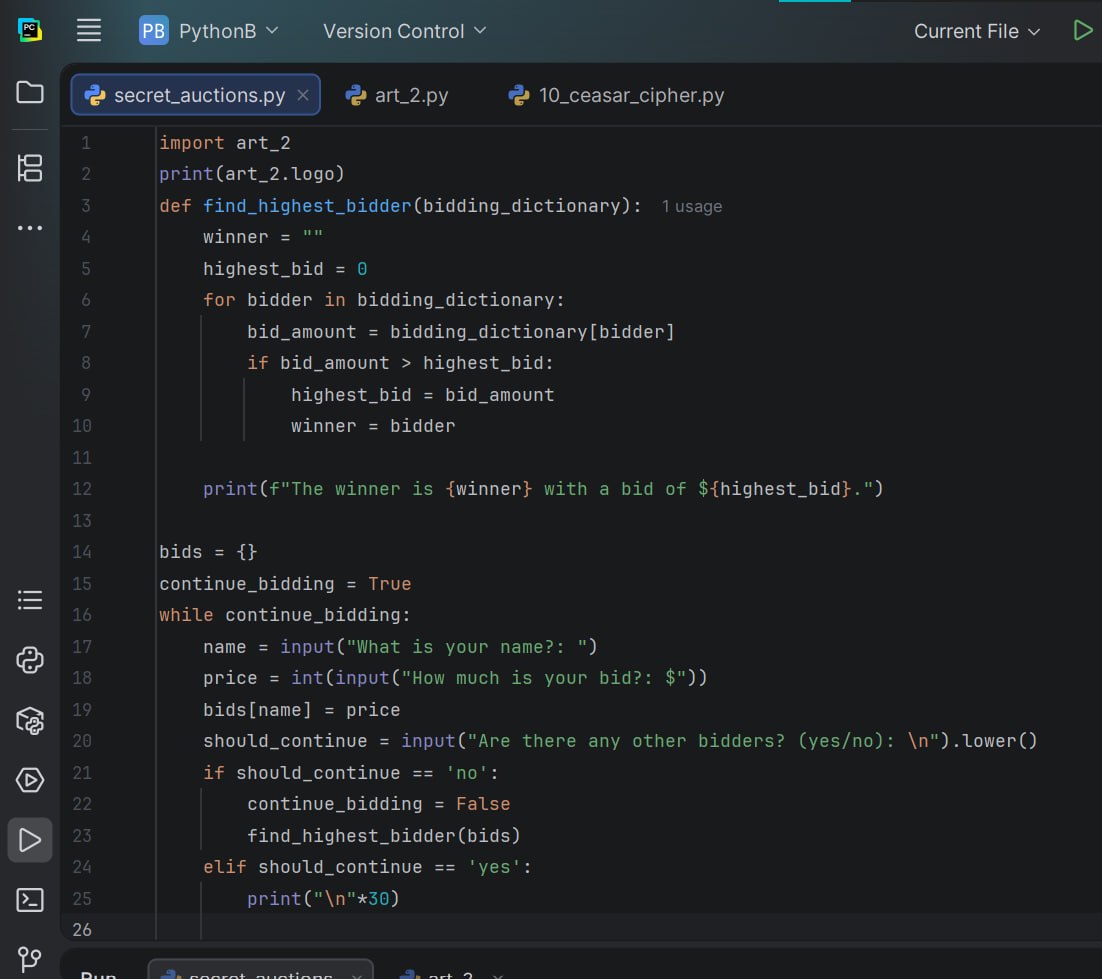
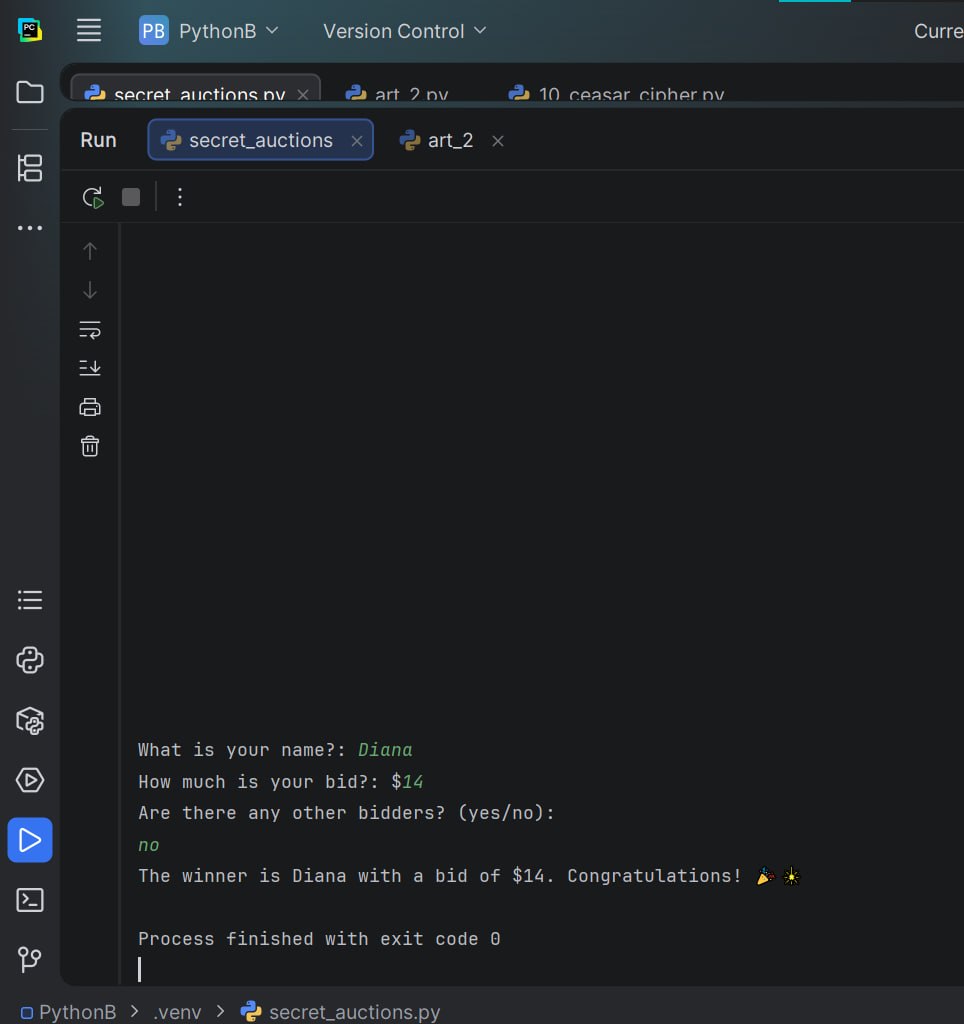
Title: Secret Auction Project
We have been working on some really challenging and exciting projects lately and learned so much from them. Really glad that I am a part of this team.
@CodeToInspire
@a_daneshyar
#AfghanGirlsCode #python #programmig #WomenInTech
1
7
96

Mar 28
Graded Quiz終了〜!
web programmigは楽勝って聞いてたけど、ふつーに初見の問題ばっかで難しかった😂
今期はBで着地やな。
まぁまぁまぁ、こんなもん。
試験おわって開放感!
週末楽しむぞ😆
#uopeople
8
531

El tutorial del framework tinyurl.com/4zvknnts llega a su 6º vídeo. Entorno #programmig #asm para #ZXSpectrum 128 usado en retrobensoft.itch.io/perseus. Facilitando el #homebrew con trucos implementados en un entorno potente, esta vez con bloques animados.
youtube.com/watch?v=2r94KFHw…
4
5
364
Clase de la asignatura "Videojuegos 2" de @UA_Universidad impartida online por @FranGallegoBR debido a ⛈️. Sistema de partículas con raylib.com/. Gestión básica de memoria con SmartPointers. Cazando bugs. #programmig #cpp
youtube.com/live/omKqLOSljmQ
1
7
295

Mar 2
The Seq programmig language
A Python dialect for bioinformatics
seq-lang.org/
Seq is a Pythonic language for computational genomics and bioinformatics. With a Python-compatible syntax and a host of domain-specific features and optimizations, Seq makes writing high-performance genomics software as easy as writing Python code, and achieves performance comparable to (and in many cases better than) C/C .
docs.seq-lang.org/
Seq starts with a subset of Python—and is in many cases a drop-in replacement—yet also incorporates novel bioinformatics- and computational genomics-oriented data types, language constructs and optimizations. Seq enables users to write high-level, Pythonic code without having to worry about low-level or domain-specific optimizations, and allows for the seamless expression of the algorithms, idioms and patterns found in many genomics or bioinformatics applications.
We evaluated Seq on several standard computational genomics tasks like reverse complementation, k-mer manipulation, sequence pattern matching and large genomic index queries. On equivalent CPython code, Seq attains a performance improvement of up to two orders of magnitude, and a 160× improvement once domain-specific language features and optimizations are used.
With parallelism, we demonstrate up to a 650× improvement. Compared to optimized C code, which is already difficult for most biologists to produce, Seq frequently attains up to a 2× improvement, and with shorter, cleaner code. Thus, Seq opens the door to an age of democratization of highly-optimized bioinformatics software.
dl.acm.org/doi/10.1145/33605…
HapTree-X
HapTree-X is a computational tool that phases various kinds of next-generation sequencing data. Currently, it supports whole-genome, whole-exome, 10X Genomics and RNA-seq data.
It is especially powerful on RNA-seq data as it can utilize allelic imbalance to better phase genic regions.
github.com/0xTCG/haptreex
HapTree-X, a probabilistic framework that utilizes latent long-range information to reconstruct unspecified haplotypes in diploid and polyploid organisms.
nature.com/articles/s41467-0…
HapTree-X's output follows the HapCUT output format convention. The output file will contain the set of phased haplotype blocks in a list format where the beginning of each block starts with BLOCK and the end of each block is indicated by *****.
github.com/vibansal/HapCUT2
HapCUT2
HapCUT2 is an iterative procedure that starts with a candidate haplotype pair. Given the current pair of haplotypes, HapCUT2 searches for a sub-set of variants (using max-cut computations in the read-haplotype graph) such that changing the phase of these variants relative to the remaining set of variants results in a new pair of haplotypes with greater likelihood.
This procedure is repeated iteratively until no further improvements can be made to the likelihood.
genome.cshlp.org/content/27/…


3
191
Mar 2
The Seq programmig language
A Python dialect for bioinformatics
seq-lang.org/
Seq is a Pythonic language for computational genomics and bioinformatics. With a Python-compatible syntax and a host of domain-specific features and optimizations, Seq makes writing high-performance genomics software as easy as writing Python code, and achieves performance comparable to (and in many cases better than) C/C .
docs.seq-lang.org/
Seq starts with a subset of Python—and is in many cases a drop-in replacement—yet also incorporates novel bioinformatics- and computational genomics-oriented data types, language constructs and optimizations. Seq enables users to write high-level, Pythonic code without having to worry about low-level or domain-specific optimizations, and allows for the seamless expression of the algorithms, idioms and patterns found in many genomics or bioinformatics applications.
We evaluated Seq on several standard computational genomics tasks like reverse complementation, k-mer manipulation, sequence pattern matching and large genomic index queries. On equivalent CPython code, Seq attains a performance improvement of up to two orders of magnitude, and a 160× improvement once domain-specific language features and optimizations are used.
With parallelism, we demonstrate up to a 650× improvement. Compared to optimized C code, which is already difficult for most biologists to produce, Seq frequently attains up to a 2× improvement, and with shorter, cleaner code. Thus, Seq opens the door to an age of democratization of highly-optimized bioinformatics software.
dl.acm.org/doi/10.1145/33605…
HapTree-X
HapTree-X is a computational tool that phases various kinds of next-generation sequencing data. Currently, it supports whole-genome, whole-exome, 10X Genomics and RNA-seq data.
It is especially powerful on RNA-seq data as it can utilize allelic imbalance to better phase genic regions.
github.com/0xTCG/haptreex
HapTree-X, a probabilistic framework that utilizes latent long-range information to reconstruct unspecified haplotypes in diploid and polyploid organisms.
nature.com/articles/s41467-0…
HapTree-X's output follows the HapCUT output format convention. The output file will contain the set of phased haplotype blocks in a list format where the beginning of each block starts with BLOCK and the end of each block is indicated by *****.
github.com/vibansal/HapCUT2
HapCUT2
HapCUT2 is an iterative procedure that starts with a candidate haplotype pair. Given the current pair of haplotypes, HapCUT2 searches for a sub-set of variants (using max-cut computations in the read-haplotype graph) such that changing the phase of these variants relative to the remaining set of variants results in a new pair of haplotypes with greater likelihood.
This procedure is repeated iteratively until no further improvements can be made to the likelihood.
genome.cshlp.org/content/27/…
4
7
136

Feb 17
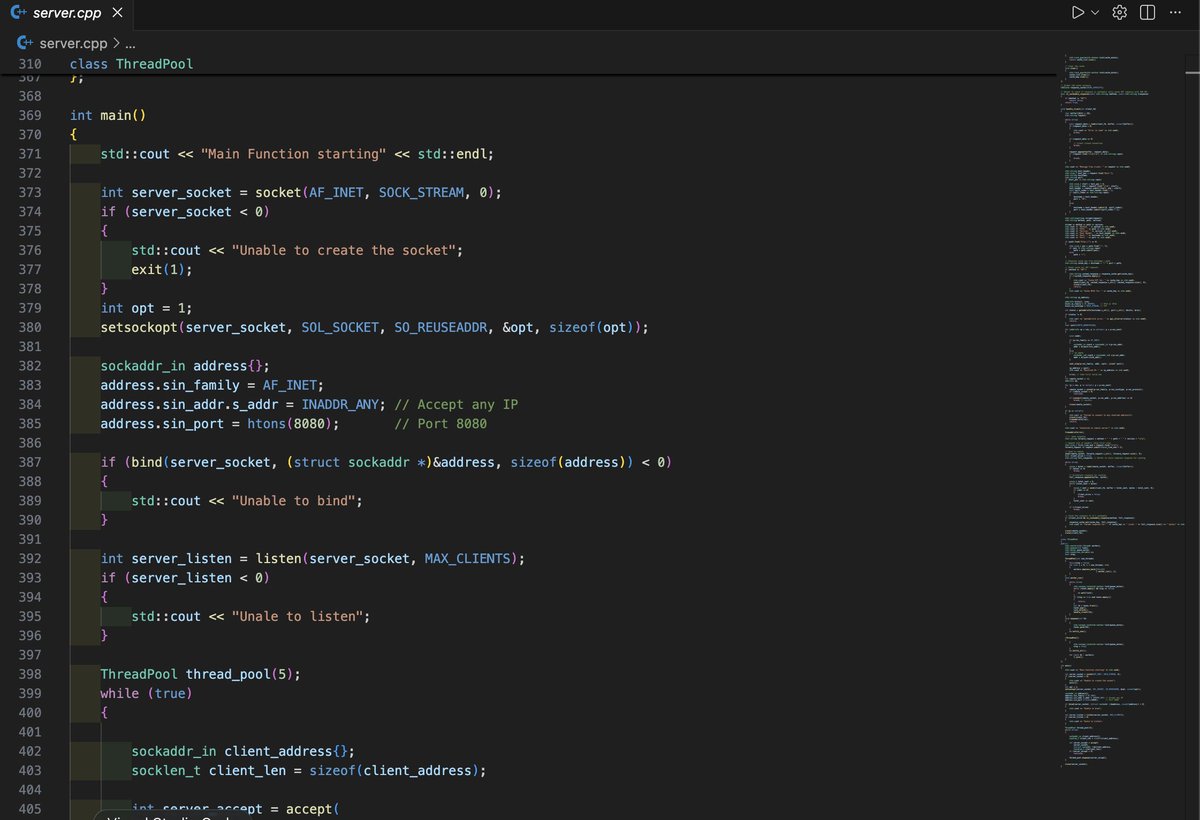
Built a multithreaded HTTP forward proxy in C .
Thread pool, LRU cache with get/put, DNS resolution, raw socket programmig, no libraries, just fundamentals.
Thanks @SinghDevHub for teaching and inspiring me
GitHub: github.com/sajalbatra/multi-…
#cpp #BuildingInPublic #learning
1
8
377


Jan 14
6. CS50's Introduction to Programming with Python.
Explore programming using Python for various applications like:
→ Data science
→ Web programmig
→ General-purpose programming
🔗 hedx.sjv.io/qz0j9y
1
10
4,031
Jan 2
6. CS50's Introduction to Programming with Python.
Explore programming using Python for various applications like:
- Data science
- Web programmig
- General-purpose programming
🔗 hedx.sjv.io/qz0j9y
1
2
85

7 Dec 2025
Yup, I know for sure DARPA was working on Neurolace Programmig with the military in 1975. Imagine how it has advaned since then. Betweee, the IEEE, MIT, MIC and Biodigital Convergence via the hijacking of our Biofields (without our inforned consent) it's now been perfected.
2
26