Molecular-level protein semantic learning via structure-aware coarse-grained language modeling
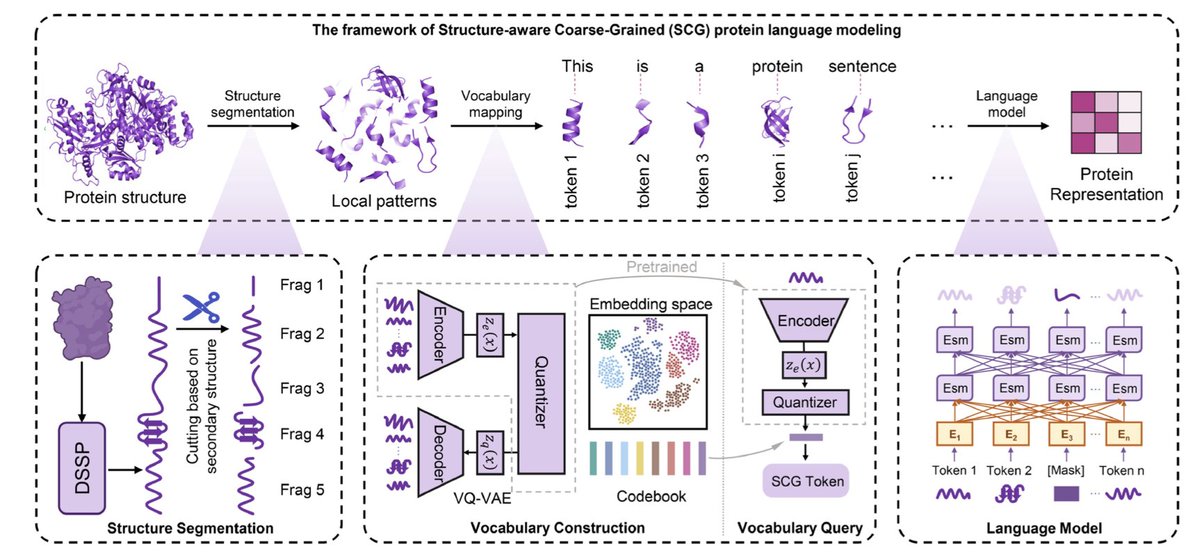
1. This study introduces a novel structure-aware coarse-grained protein language that redefines protein representation by integrating secondary structure segmentation with vector quantization techniques. The framework partitions proteins into biologically meaningful structural fragments, significantly reducing sequence length while preserving functional semantics.
2. The proposed SCG (Structure-Aware Coarse-Grained) language outperforms traditional amino acid sequences and state-of-the-art protein languages in capturing molecular-level semantics, especially for long proteins. This is achieved by constructing a compact vocabulary of local structural patterns derived from secondary structures.
3. The SCG framework mitigates semantic truncation in large proteins during language model processing. Empirical evaluations show that SCG representations experience minimal truncation effects compared to fine-grained languages, preserving structural and functional context in downstream tasks.
4. The study explores different segmentation strategies and confirms that secondary structure-based segmentation is essential for preserving biologically relevant features. Alternative segmentation schemes like uniform random and dynamic random fragment segmentation lead to significant performance degradation.
5. The SCG method employs a multi-head attention encoder in VQ-VAE to improve codebook utilization and vocabulary construction. This design enhances stability and efficiency in vector quantization, outperforming traditional clustering methods like k-means.
6. The SCG language demonstrates stable performance across various downstream tasks, including function prediction, enzyme classification, and interaction identification. It shows consistent advantages when applied to both lightweight and deep language models.
7. The study highlights the potential of combining SCG representations with existing protein modeling approaches to further improve modeling efficiency and scalability. Future work may explore extending this coarse-grained paradigm to graph neural networks for scalable, structure-aware protein modeling.
8. The authors have made the data and source code available on GitHub and Zenodo, facilitating reproducibility and further research in the field.
💻Code: github.com/bug-0x3f/coarse-g…
📜Paper: doi.org/10.1093/bioinformati…
#ProteinLanguageModeling #CoarseGrainedRepresentation #StructuralBioinformatics #Bioinformatics #ProteinFunctionPrediction
4
15
1,196

4 Aug 2025
Scaling down protein language modeling with MSA Pairformer
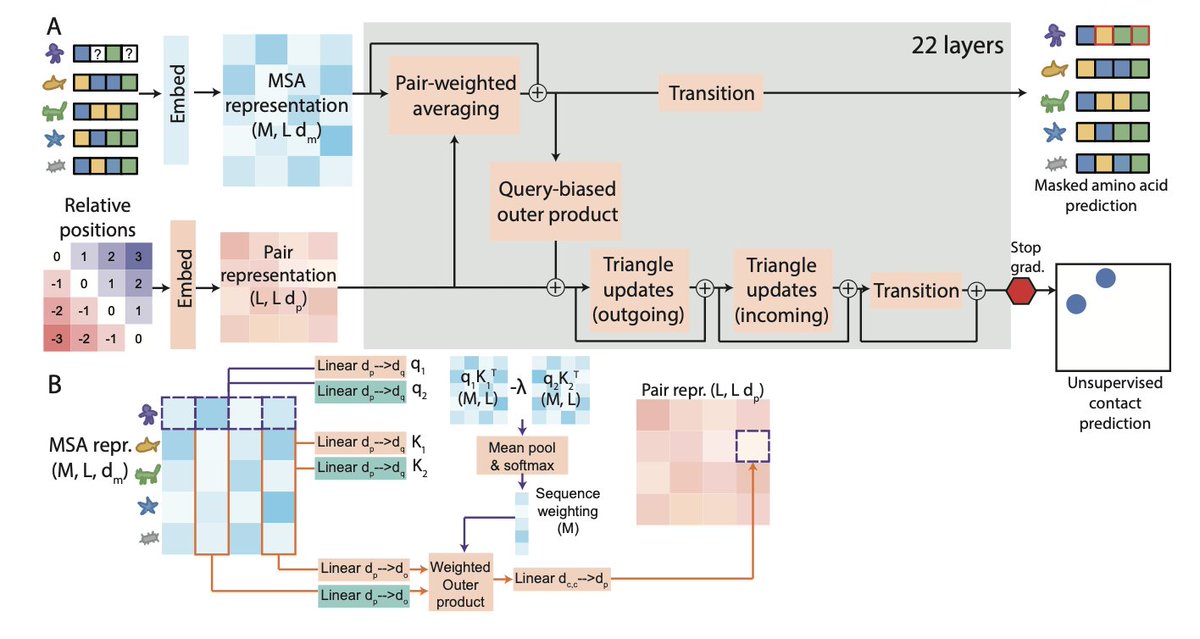
1. The article introduces MSA Pairformer, a novel protein language model that achieves state-of-the-art performance with significantly fewer parameters compared to existing models. MSA Pairformer uses a memory-efficient architecture to process multiple sequence alignments (MSAs) and extract evolutionary signals relevant to a query sequence.
2. A key innovation of MSA Pairformer is its query-biased outer product operation, which selectively weights sequences based on their evolutionary relevance to the query sequence. This allows the model to capture subfamily-specific co-evolutionary signals within large protein families, addressing a limitation of previous MSA models.
3. MSA Pairformer demonstrates superior performance in unsupervised contact prediction, outperforming models like ESM2-15B by 6% points while using two orders of magnitude fewer parameters. It also shows substantial improvements in predicting contacts at protein-protein interfaces, with a 24% point increase over MSA Transformer.
4. Unlike single-sequence models that struggle with variant effect prediction as they scale, MSA Pairformer maintains strong performance in both contact prediction and zero-shot variant effect prediction. This highlights its ability to balance evolutionary signal extraction and functional prediction.
5. Ablation studies reveal that triangle operations in MSA Pairformer help remove indirect correlations between residues, enabling more accurate contact predictions. Additionally, MSA Pairformer does not hallucinate contacts after removing covariance from MSAs, unlike MSA Transformer.
6. The model’s ability to extract subfamily-specific signals and its robustness to MSA perturbations open new avenues for biological discovery, including the potential to explore alternative conformations and interactions within protein families.
7. MSA Pairformer challenges the current scaling paradigm in protein language modeling by demonstrating that parameter efficiency and biological insight can synergistically advance the field. It enables efficient adaptation to rapidly expanding sequence databases and paves the way for more sustainable and scalable protein language models.
@yoakiyama @ZhidianZ @sokrypton
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/yoakiyama/MSA_Pai…
#ProteinLanguageModeling #MSAPairformer #Bioinformatics #ProteinStructurePrediction #MachineLearning
5
29
2,026