MZSGO: Multimodal zero-shot protein function annotation via evolutionary signals and textual semantics
1. MZSGO reframes protein function prediction as a Protein–GO matching task, enabling true zero-shot inference for GO terms never seen during training by comparing protein evidence to GO term definitions in a shared embedding space.
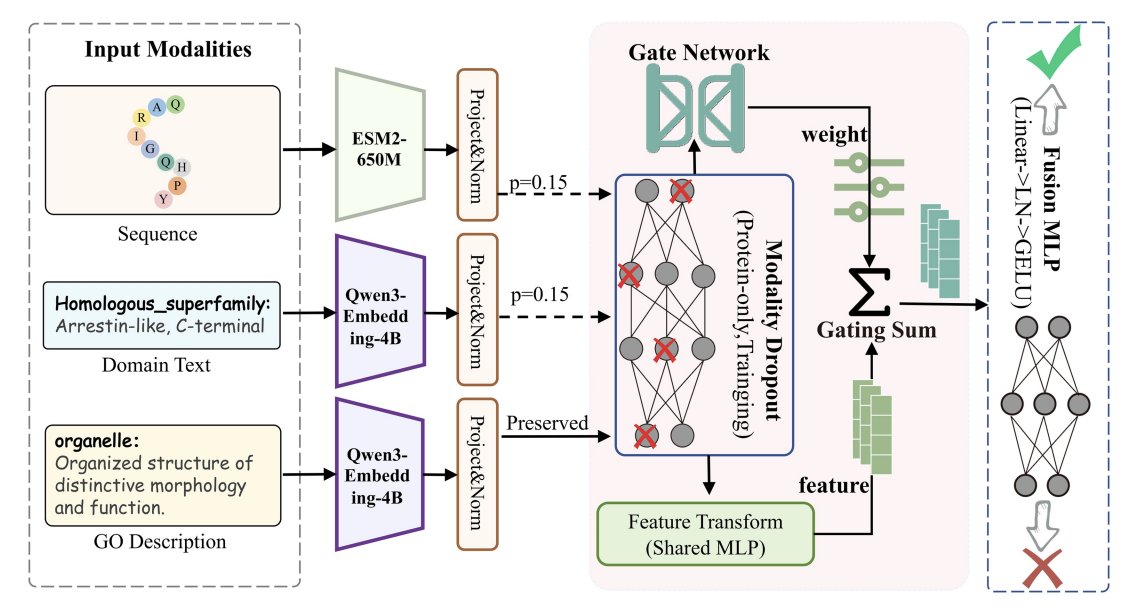
2. The key multimodal idea: combine evolutionary sequence signals (ESM2-650M embeddings) with textual semantics from (a) protein domain descriptions and (b) GO term definitions, both encoded by a unified text embedding model (Qwen3-Embedding-4B).
3. Instead of treating domains and labels as categorical IDs, MZSGO uses their natural-language descriptions/definitions as semantic anchors—so “what a GO term means” becomes directly learnable and transferable to new/rare labels.
4. Architecture highlights: (i) modality-specific projection MLPs to a shared latent space, (ii) asymmetric modality dropout that randomly removes sequence or domain features during training (but keeps GO text), and (iii) label-aware adaptive gated fusion to weight sequence/domain/label signals per protein–GO pair.
5. Why asymmetric dropout matters: it simulates real annotation settings where protein-derived evidence (domains) can be missing or incomplete, while GO term text is always available at inference—improving robustness and reducing reliance on any single protein modality.
6. Why gated fusion matters: naive concatenation can inject noise when modalities contribute unevenly; the learned gate assigns context-dependent weights (sequence vs domain vs GO definition), improving both supervised accuracy and especially generalization to unseen labels.
7. Dataset design targets realistic zero-shot evaluation: training uses CAFA5 Swiss-Prot with GO version cutoff (Jan 2023), while zero-shot labels are defined by a later GO release (Oct 2025). Test proteins are homology-filtered (Diamond, remove >30% identity to training) to prevent leakage.
8. Performance summary (standard test set): MZSGO is competitive on supervised metrics (Fmax/AUPR) and stands out on unseen-label generalization. Example: BP Unseen AUPR 0.2393 vs ProtNote 0.0303; MF Unseen AUPR 0.4806; CC Unseen AUPR 0.5862. Harmonic mean (seen/unseen) also improves strongly (BP 0.3241, MF 0.5821, CC 0.6669).
9. True temporal zero-shot benchmark (new GO terms added after training cutoff): MZSGO improves both precision and overall reliability vs ProtNote—e.g., CC Fmax 0.6610 vs 0.2983, with much higher precision (0.5521 vs 0.1853), suggesting fewer text-driven false positives.
10. Ablations indicate zero-shot transfer depends on semantic domain text (not one-hot domains), and on adaptive fusion modality dropout. Removing dropout can slightly help seen-label Fmax but hurts zero-shot balance; replacing gated fusion with concatenation reduces the seen/unseen trade-off.
💻Code: github.com/toxic-byte/MZSGO
📜Paper: doi.org/10.1093/bioinformati…
#ProteinFunctionPrediction #GeneOntology #ZeroShotLearning #ProteinLanguageModels #LLM #MultimodalAI #ComputationalBiology #Bioinformatics #CAFA #SwissProt
1
8
915
LAFA: A Framework for Reproducible Longitudinal Assessment of Protein Function Annotation Models
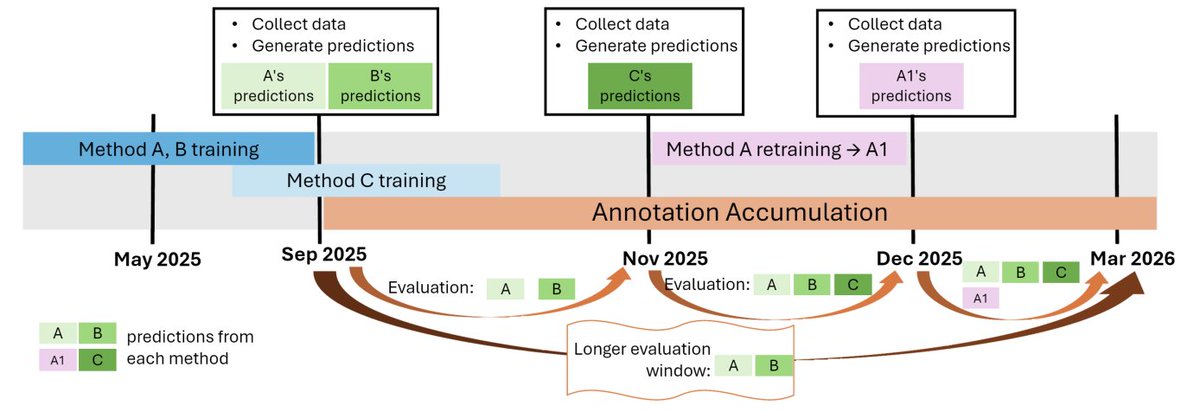
1 LAFA is introduced as a persistent, CAFA-style benchmarking server that continuously evaluates protein function prediction methods as Gene Ontology (GO) annotations accumulate, rather than waiting for triennial CAFA rounds.
2 The key motivation is the “open world” issue in function annotation: during a fixed CAFA accumulation window, correct predictions may not yet be experimentally validated, so methods can be under-credited; LAFA re-evaluates over evolving ground truth to track performance more fairly through time.
3 LAFA emphasizes reproducibility and usability by hosting methods as offline containers (packaging code dependencies), helping ensure that assessments remain runnable over time and reducing leakage by preventing access to post–t0 data during evaluation.
4 The system mirrors CAFA’s prospective logic with a streamlined timeline: at each UniProt-GOA release time point (t0), targets are defined and hosted methods generate predictions immediately; evaluations are then computed for multiple “time windows” (t0→t1) as new releases arrive.
5 The backend is implemented as two reusable Nextflow workflows: a “time point build” workflow (data acquisition preprocessing prediction generation) and a “time window evaluation” workflow (constructing ground truth from newly accumulated experimental annotations and scoring methods via CAFA-evaluator).
6 Data sources include UniProt-GOA (annotations), UniProtKB (sequences), and the GO Data Archive (ontology graph). LAFA uses SwissProt (reviewed UniProt) as the intended stable test bed (≈550k–580k proteins historically), and also constructs a high-quality training set from experimentally validated annotations.
7 For the current testbed stage (compute-limited), the paper reports retrospective runs on a smaller “ground truth target set” of 7,401 proteins whose sequences remained unchanged and gained experimental annotations between the 09/2025 and 03/2026 UniProt-GOA releases; the authors plan scaling to full SwissProt.
8 Initial hosted containerized methods include TransFew, FunBind (sequence-only version of a multimodal foundation model), and DeepGOPlus, alongside four baselines (Naive frequency, Non-experimental GOA carryover, BLAST transfer, and embedding-similarity transfer). Retrained model versions can be submitted and compared longitudinally to quantify training-data recency effects.
9 The front-end website provides interactive browsing of results across time windows, including GO aspect–specific views (MF/BP/CC), best-threshold F1 summaries, and precision–recall curves; it also supports direct comparisons between two time windows to study robustness vs accumulation length and performance decay over time.
10 Future directions highlighted include partnering with UniProt-GOA for a private hold-out of newly curated annotations to enable faster evaluation of newly arriving methods, scaling compute for full SwissProt prediction generation, and adding additional metrics (e.g., GO-slim-based evaluation) to improve interpretability and reduce sensitivity to ontology artifacts.
💻Code: github.com/anphan0828/CAFA_f…
📜Paper: arxiv.org/abs/2604.20782
#ProteinFunctionPrediction #GeneOntology #Benchmarking #Reproducibility #Bioinformatics #ComputationalBiology #MachineLearning #Workflows #Nextflow #Containers
2
817
STAR-GO: Improving protein function prediction by learning to hierarchically integrate ontology-informed semantic embeddings
1. STAR-GO targets a practical failure mode in protein function prediction: Gene Ontology (GO) keeps evolving, so models trained on a fixed GO snapshot struggle with unseen/new GO terms. STAR-GO is designed for strong zero-shot generalization while staying competitive in standard supervised benchmarks.
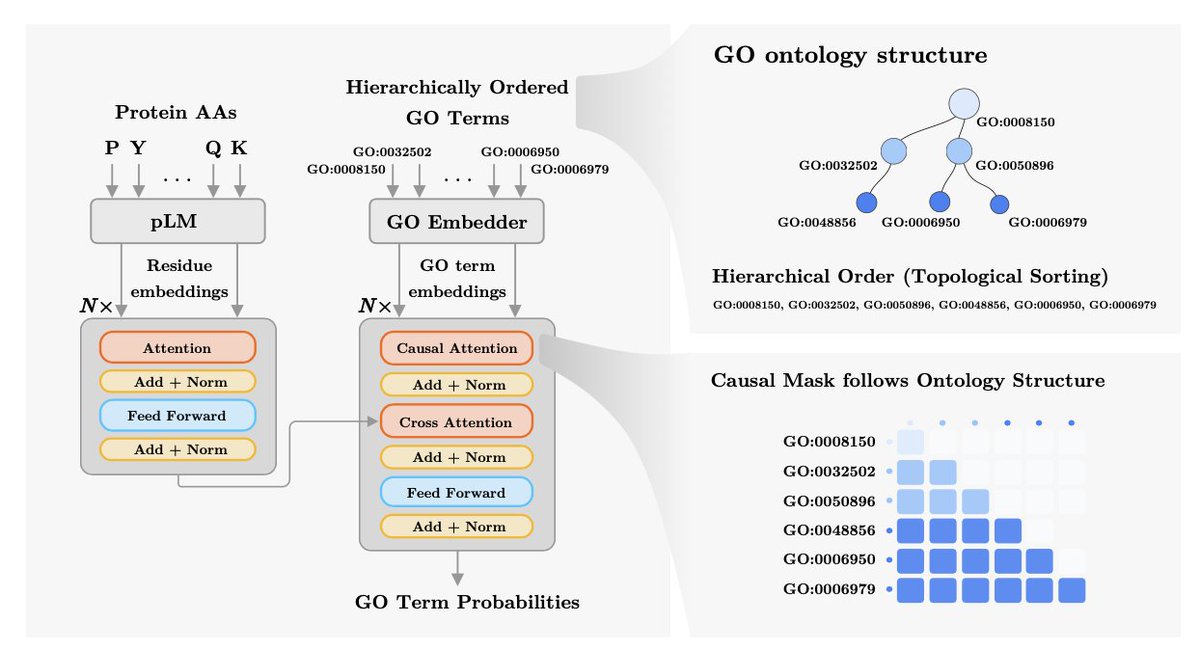
2. Core idea: learn GO term representations that jointly capture (a) term-definition semantics from biomedical language models and (b) GO graph structure, then decode GO terms in hierarchical order so information can flow from general ancestors to specific descendants.
3. GO embedding module: each GO term definition is embedded with SBERT-BioBERT (sentence-level, mean pooled), then passed through a trainable projection autoencoder that injects structure via multi-task supervision—predicting the term’s ancestor set, sub-ontology (MF/BP/CC), and term identity.
4. Why this matters for zero-shot: description-based GO embeddings preserve semantic similarity even when the GO graph doesn’t explicitly connect terms tightly (example discussed: ATP binding vs ATP hydrolysis activity). Unlike one-hot/graph-only embeddings, STAR-GO can embed a newly introduced GO term directly from its text definition.
5. Model architecture: an encoder–decoder Transformer. The encoder ingests frozen residue embeddings from ProtT5 and contextualizes them with self-attention. The decoder takes GO term embeddings and uses (i) causal self-attention plus a mask aligned to a topological (ancestor→descendant) GO ordering and (ii) cross-attention over protein residues to compute term probabilities.
6. Hierarchical decoding is an explicit inductive bias: the decoder’s causal mask ensures each GO term can attend to earlier (more general / ancestral) terms, encouraging consistent propagation across ontology levels rather than treating labels as independent.
7. Standard setting results (DeepFRI-derived dataset, 36,641 proteins; 2,752 GO terms): STAR-GO is competitive with recent methods and shows particularly strong term-level discrimination (best AUC across BP/CC/MF: BP 0.989, CC 0.988, MF 0.995), while maintaining solid Fmax (BP 0.548, CC 0.659, MF 0.719).
8. Zero-shot evaluation (DeepGOZero protocol; 16 held-out GO terms): STAR variants achieve the best zero-shot AUC in 13/16 cases versus DeepGOZero, DeepGO-SE, and TransFew. Text-only embeddings often win on MF/BP terms, structure-only wins on a few cases, and the combined model is the most stable across ontologies.
9. Interpretability in the zero-shot setting: cross-attention rollout highlights residues relevant to unseen GO terms. For held-out DNA-binding transcription factor terms (GO:0001228 activator, GO:0001227 repressor), attention-derived residue scores align with experimentally curated DNA-contact sites (BioLiP), with binding-site recovery AUROC up to 0.841 on tested PDB chains—despite the model never seeing those GO terms during training.
10. Ablations clarify what drives performance: removing ontology-informed GO embeddings or removing hierarchical ordering degrades results; MLP-style fusion underperforms the decoder with hierarchical ordering; ProtT5 residue embeddings outperform ESM-1b/ESM-2 within this framework, especially on MF Macro AUPR and Fmax.
💻Code: github.com/boun-tabi-lifelu/…
📜Paper: arxiv.org/abs/2512.05245
#ProteinFunctionPrediction #GeneOntology #ZeroShotLearning #Transformers #ProteinLanguageModels #Bioinformatics #ComputationalBiology #MachineLearning
14
1,673
STAR-GO: Improving Protein Function Prediction by Learning to Hierarchically Integrate Ontology-Informed Semantic Embeddings
1. STAR-GO targets a practical pain point in protein function prediction: GO keeps evolving, so models trained on a fixed label set become outdated. It addresses this with strong zero-shot prediction by building GO term representations that can be generated from term definitions at inference time (no retraining needed when new terms appear).
2. The key idea is to jointly model two complementary signals in Gene Ontology terms: (a) semantics from textual definitions and (b) structure from the GO DAG. STAR-GO learns unified, ontology-informed GO embeddings and then aligns them with protein sequence embeddings to predict functions.
3. GO embedding module: each GO term definition is embedded using SBERT-BioBERT sentence embeddings, then refined by a projection autoencoder trained with multi-task supervision to recover (i) ancestor terms, (ii) sub-ontology (MF/BP/CC), and (iii) term identity. This “structure-recovering” training injects hierarchy while preserving semantic similarity from text.
4. Model architecture: an encoder–decoder Transformer fuses residue-level protein embeddings (from ProtT5) with GO term embeddings. The decoder uses cross-attention so each GO term can attend to informative residues, learning sequence–function associations in a label-aware way rather than treating labels as independent.
5. Hierarchical decoding is an explicit inductive bias: GO terms are topologically sorted (ancestors to descendants) and decoded with causal self-attention, enabling information flow from general functions to more specific ones. This helps capture dependencies across ontology levels and supports consistent predictions under the GO hierarchy.
6. Standard supervised evaluation (DeepFRI-derived dataset; 36,641 proteins; 2,752 GO terms): STAR-GO is competitive with strong baselines (PFresGO, DeepFRI, TALE , DeepGOZero, DeepGO-SE, TransFew). It achieves particularly strong term-level discriminability, reporting top AUC across BP/CC/MF (e.g., MF AUC 0.995; CC AUC 0.988; BP AUC 0.989).
7. Zero-shot evaluation (DeepGOZero protocol; 16 held-out GO terms across MF/BP/CC): STAR-GO variants achieve the best AUC in 13/16 held-out terms vs DeepGOZero, DeepGO-SE, and TransFew. Text-only GO embeddings (definitions) often dominate in zero-shot MF/BP, while structure-only can win on select terms—supporting the claim that semantics and structure contribute differently depending on the term.
8. Ablations clarify what matters: (i) removing either semantic or structural GO information reduces performance; (ii) flattening GO term ordering hurts, confirming the value of hierarchical decoding; (iii) training GO embeddings from scratch is notably worse, showing the benefit of ontology-informed pretrained GO representations.
9. Interpretability in a true zero-shot setting: cross-attention rollout highlights residues relevant to unseen DNA-binding transcription factor terms (GO:0001228 activator, GO:0001227 repressor). Attention-derived residue scores align with BioLiP DNA-contact sites, with binding-site recovery AUROC up to 0.841—suggesting the model learns transferable mappings from GO semantics to residue-level signals.
💻Code: github.com/boun-tabi-lifelu/…
📜Paper: arxiv.org/abs/2512.05245
#ComputationalBiology #Bioinformatics #ProteinFunctionPrediction #GeneOntology #Transformers #ZeroShotLearning #ProteinLanguageModels #RepresentationLearning

3
18
1,287
Molecular-level protein semantic learning via structure-aware coarse-grained language modeling
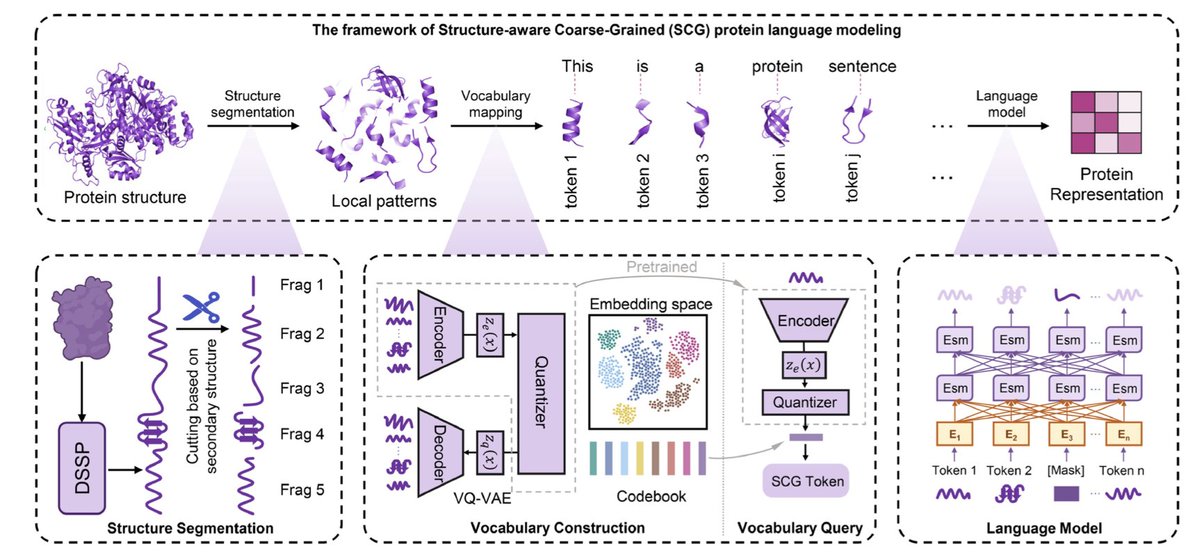
1. This study introduces a novel structure-aware coarse-grained protein language that redefines protein representation by integrating secondary structure segmentation with vector quantization techniques. The framework partitions proteins into biologically meaningful structural fragments, significantly reducing sequence length while preserving functional semantics.
2. The proposed SCG (Structure-Aware Coarse-Grained) language outperforms traditional amino acid sequences and state-of-the-art protein languages in capturing molecular-level semantics, especially for long proteins. This is achieved by constructing a compact vocabulary of local structural patterns derived from secondary structures.
3. The SCG framework mitigates semantic truncation in large proteins during language model processing. Empirical evaluations show that SCG representations experience minimal truncation effects compared to fine-grained languages, preserving structural and functional context in downstream tasks.
4. The study explores different segmentation strategies and confirms that secondary structure-based segmentation is essential for preserving biologically relevant features. Alternative segmentation schemes like uniform random and dynamic random fragment segmentation lead to significant performance degradation.
5. The SCG method employs a multi-head attention encoder in VQ-VAE to improve codebook utilization and vocabulary construction. This design enhances stability and efficiency in vector quantization, outperforming traditional clustering methods like k-means.
6. The SCG language demonstrates stable performance across various downstream tasks, including function prediction, enzyme classification, and interaction identification. It shows consistent advantages when applied to both lightweight and deep language models.
7. The study highlights the potential of combining SCG representations with existing protein modeling approaches to further improve modeling efficiency and scalability. Future work may explore extending this coarse-grained paradigm to graph neural networks for scalable, structure-aware protein modeling.
8. The authors have made the data and source code available on GitHub and Zenodo, facilitating reproducibility and further research in the field.
💻Code: github.com/bug-0x3f/coarse-g…
📜Paper: doi.org/10.1093/bioinformati…
#ProteinLanguageModeling #CoarseGrainedRepresentation #StructuralBioinformatics #Bioinformatics #ProteinFunctionPrediction
4
15
1,196

22 Nov 2025
Integrating Millions of Years of Evolutionary Information into Protein Structure Models for Function Prediction
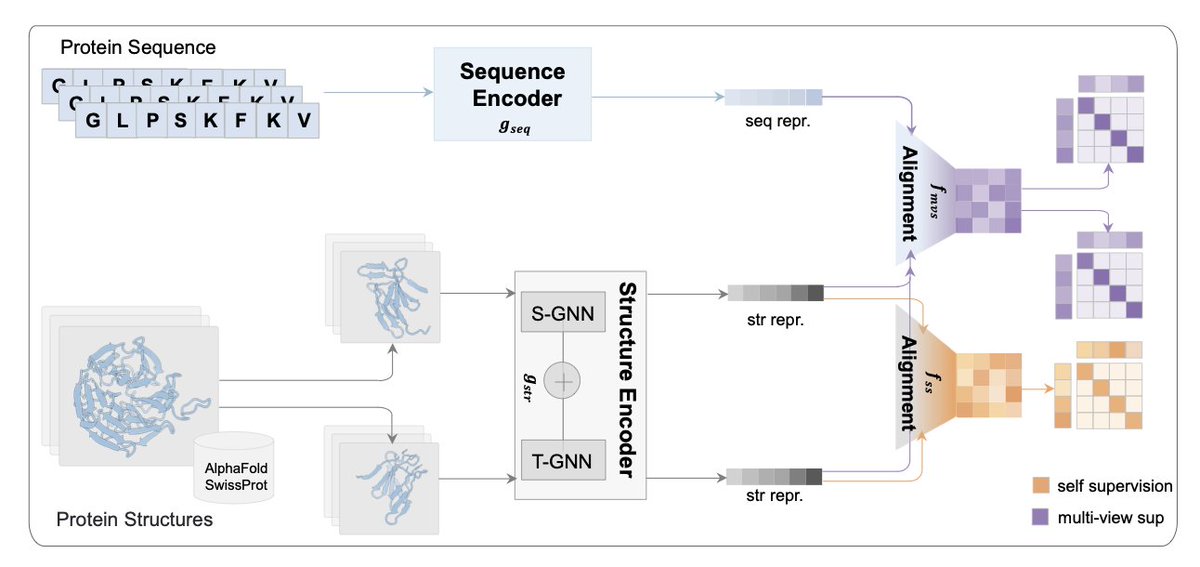
1. Researchers have developed a novel framework called ESMSCOP that integrates evolutionary information from protein sequences with detailed 3D structural data to improve protein function prediction. This approach leverages contrastive learning to bridge the gap between sequence and structure, capturing both evolutionary and spatial insights.
2. The study introduces a multi-view encoder architecture that jointly learns from sequence and structure modalities. By incorporating a state-of-the-art protein language model (ESM-C), ESMSCOP effectively distills millions of years of evolutionary context into protein representations, enhancing the accuracy of function prediction tasks.
3. ESMSCOP employs a contrastive pre-training strategy with self-supervision and multi-view supervision. This allows the model to align sequence and structure representations, enforcing consistency across different structural conformations and improving the robustness of learned embeddings.
4. Extensive experiments on multiple benchmark datasets demonstrate that ESMSCOP outperforms existing methods in protein function prediction, even when using relatively less pre-training data. The model shows strong performance across diverse tasks, including enzyme classification and protein active site prediction.
5. The framework is designed to work under limited label availability, making it applicable to low-resource settings. This is particularly important given the scarcity of annotated protein data, which often hinders deep learning approaches in bioinformatics.
6. Future work will focus on improving the structure-based encoder using E(3)-equivariant models and further exploring the integration of pre-trained protein language models with structural models to enhance performance. This research could accelerate the development of new drugs by providing more accurate protein function predictions.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinFunctionPrediction #EvolutionaryInformation #ContrastiveLearning #Bioinformatics #DeepLearning
4
17
1,566
28 Oct 2025
From Signal to Symphony: Exploring 2D Sequence Representations for Protein Function Prediction
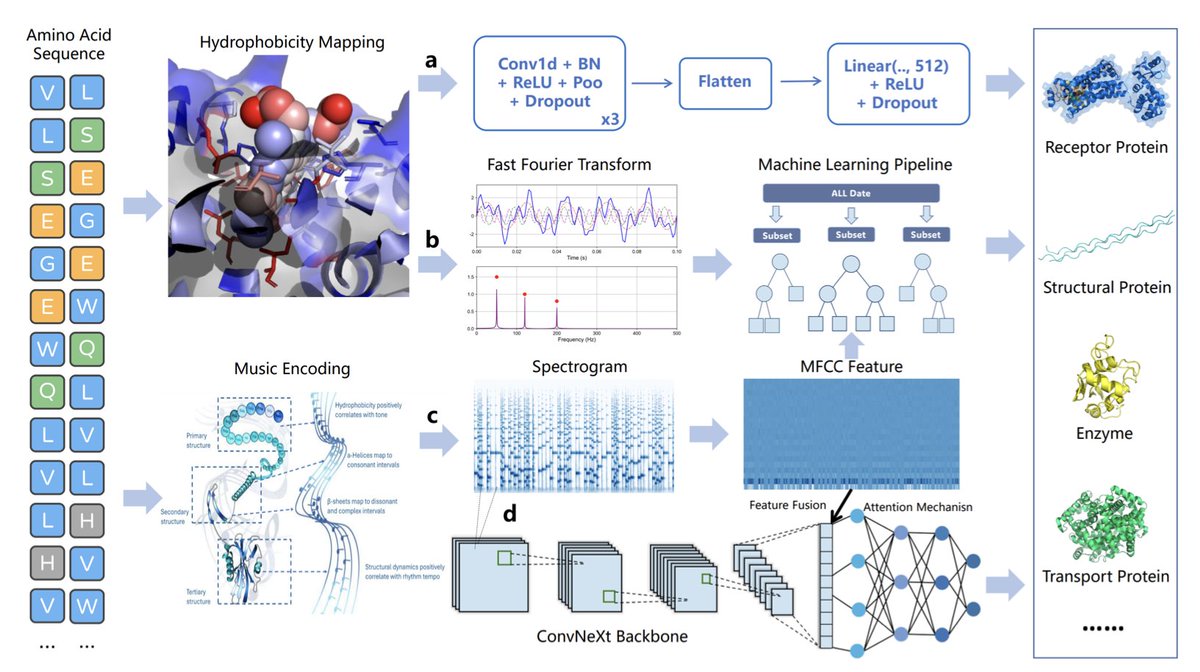
1. This study introduces a novel approach to protein function prediction by converting amino acid sequences into 2D spectrograms through a process called protein sonification. The authors developed a benchmark dataset of 18,000 sequences across 12 diverse protein classes to systematically evaluate this method.
2. The core innovation lies in the transformation of 1D sequence data into a 2D representation, which allows models to capture long-range dependencies and complex patterns more effectively. This structural change is shown to be a major contributor to the model's predictive performance.
3. The authors conducted ablation studies to determine whether the performance boost comes from the specific biophysical encoding rules or the 2D representation itself. Surprisingly, models using purely visual or acoustic features from the spectrogram achieved high accuracy, indicating that the representation itself is key.
4. The biophysically informed model achieved an accuracy of 84.00%, demonstrating that incorporating domain knowledge can provide an additional performance benefit. When trained from scratch, their fusion model outperformed standard transformer architectures like ESM-2 and ProtBERT on their dataset, highlighting its data efficiency.
5. The study also validated the generalizability of their approach on an external enzyme classification benchmark, achieving 90.44% accuracy. Additionally, they explored using their encoding to guide a diffusion model in generating novel GFP variants, which were computationally assessed for structural viability.
6. The authors conclude that the primary benefit of sonification stems from the structural transformation into a 2D format, which enables the capture of emergent functional patterns. This work opens new avenues for feature engineering in biological sequence analysis and demonstrates potential for both predictive and generative protein modeling.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinFunctionPrediction #Sonification #Bioinformatics #DeepLearning #ProteinDesign
3
14
1,327
28 Oct 2025
A Novel Framework for Multi-Modal Protein Representation Learning
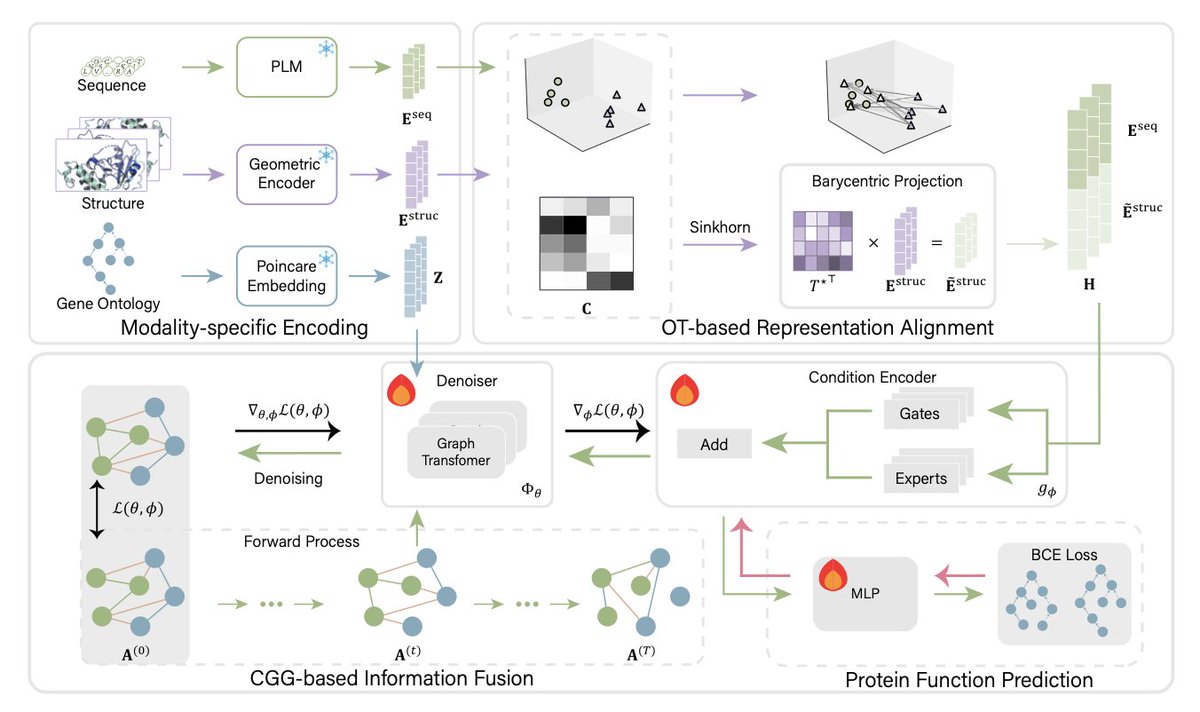
1. The article introduces DAMPE, a new framework for protein function prediction that integrates sequence, structure, and extrinsic data like protein-protein interactions and Gene Ontology annotations. This integration is crucial for accurate function prediction as it combines intrinsic and extrinsic biological signals.
2. DAMPE addresses two key challenges: cross-modal distributional mismatch and noisy relational graphs. It uses Optimal Transport (OT) for representation alignment, effectively mitigating differences between sequence and structure embeddings, and Conditional Graph Generation (CGG) for robust information fusion, avoiding issues with noisy PPI networks.
3. The framework demonstrates significant improvements over state-of-the-art methods like DPFunc, achieving AUPR gains of 0.002–0.013 and Fmax gains of 0.004–0.007. Ablation studies highlight the contributions of both OT and CGG components, with OT contributing 0.043–0.064pp AUPR and CGG adding 0.005–0.111pp Fmax.
4. Theoretical analysis in the paper shows that the CGG objective drives the condition encoder to absorb graph-aware knowledge into protein representations, enhancing the robustness and effectiveness of the embeddings for downstream tasks.
5. DAMPE offers a scalable and theoretically grounded approach for multi-modal protein representation learning. It avoids the computational overhead of traditional GNNs by using a lightweight Mixture-of-Experts architecture, making it suitable for large-scale applications.
6. The framework is evaluated on standard Gene Ontology benchmarks, demonstrating superior performance across multiple metrics. It effectively captures functional patterns in protein embeddings, as shown through qualitative evaluations and clustering metrics.
📜Paper: arxiv.org/abs/2510.23273v1
#ProteinFunctionPrediction #MultiModalLearning #OptimalTransport #ConditionalGraphGeneration #Bioinformatics #DeepLearning
2
26
2,031

17 Oct 2025
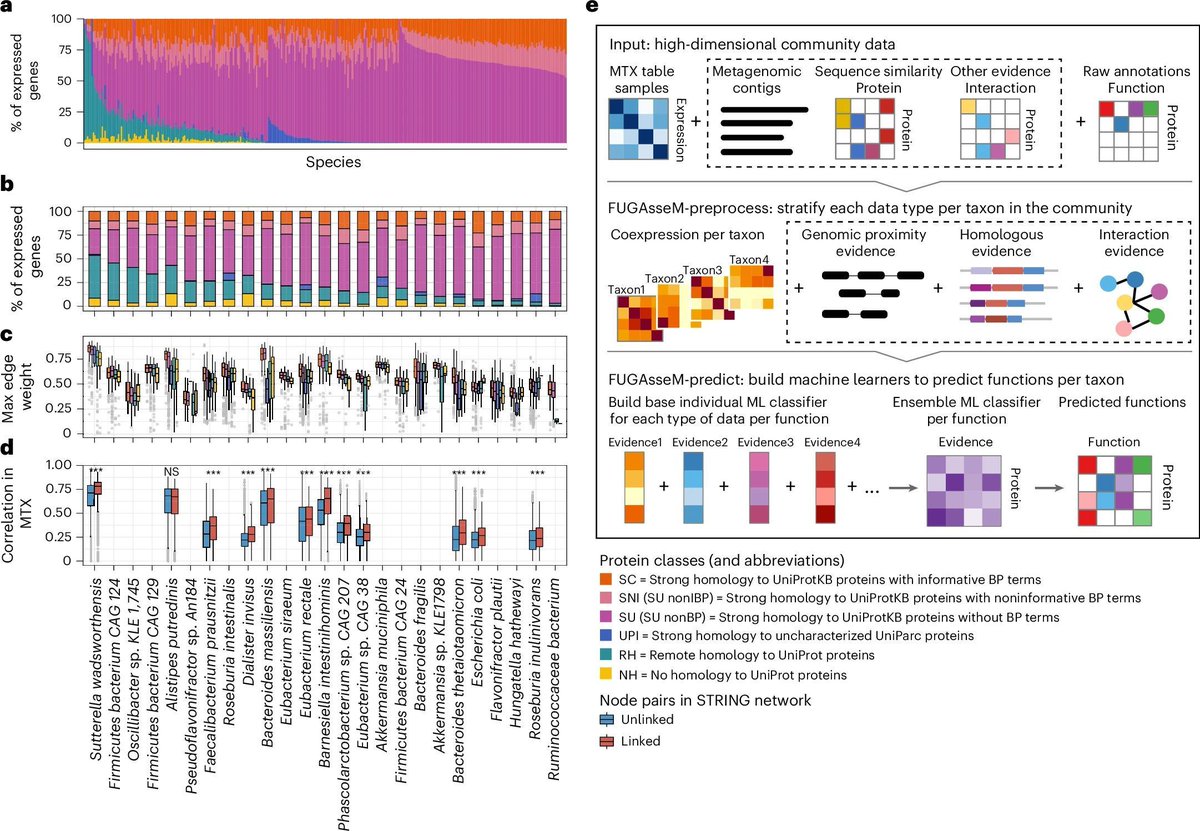
Predicting functions of uncharacterized gene products from microbial communities. #Metagenomics #GutMetagenomes #GutMetatranscriptomes #ProteinFunctionPrediction #MicrobialCommunities #Genomics #Bioinformatics @NatureBiotech
nature.com/articles/s41587-0…
1
6
390
15 Oct 2025
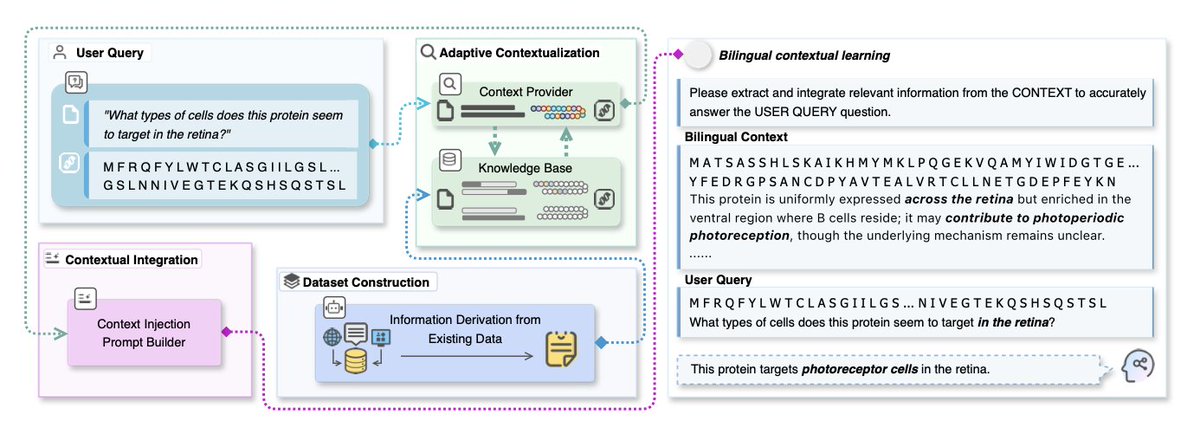
Protein as a Second Language for LLMs
1. A novel study has introduced a novel framework that enables large language models (LLMs) to interpret protein sequences as a symbolic language, significantly enhancing their ability to understand and predict protein functions without task-specific fine-tuning.
2. The core innovation lies in treating protein sequences as a "second language" that LLMs can learn through contextual examples. This approach constructs sequence–question–answer triples that reveal functional cues, allowing LLMs to acquire protein semantics in a zero-shot setting.
3. The researchers curated a bilingual dataset of 79,926 protein–QA instances, covering attribute prediction, descriptive understanding, and extended reasoning. This dataset supports effective learning and benchmarking for protein language models.
4. Empirical results show that this method delivers consistent gains across diverse open-source LLMs and GPT-4o, achieving up to 17.2% ROUGE-L improvement and even surpassing fine-tuned protein-specific language models.
5. The adaptive context construction mechanism selectively builds bilingual learning contexts for each query, exposing LLMs to new words in context so that meaning and usage can be inferred, similar to how humans acquire a second language.
6. The study demonstrates that general-purpose LLMs, when guided with protein-as-language cues, can outperform domain-specialized models, offering a scalable pathway for protein understanding.
7. The framework has been evaluated on multiple protein-language datasets, showing significant improvements over zero-shot baselines and highlighting the effectiveness of context-driven learning in bridging protein sequences with functional descriptions.
📜Paper: arxiv.org/abs/2510.11188v1
#ProteinLanguage #LLMs #Bioinformatics #AIinBiology #ProteinFunctionPrediction
2
17
1,613
8 Oct 2025
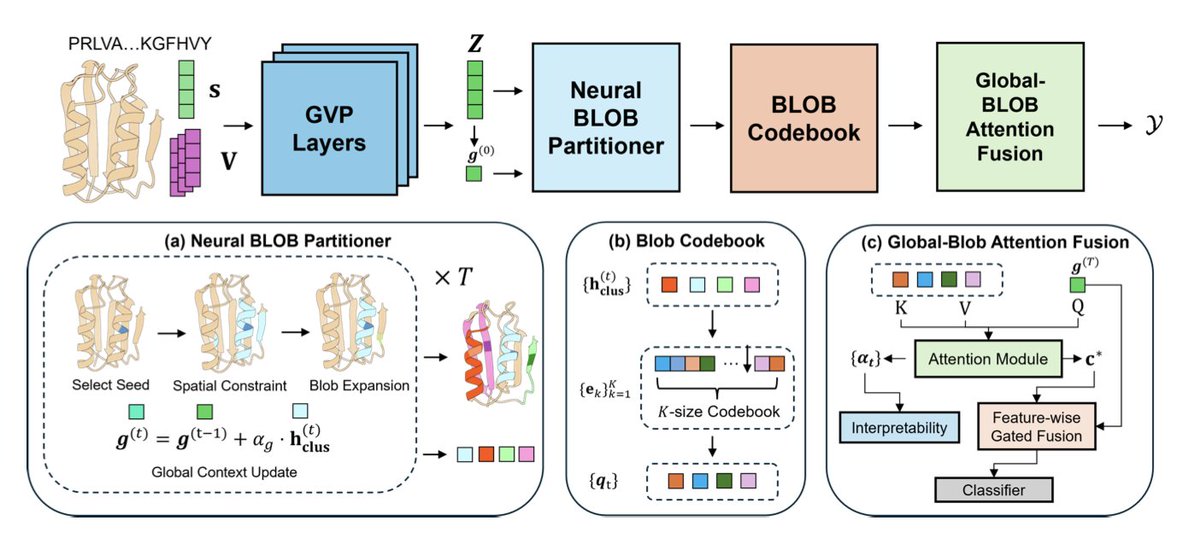
BIOBLOBS: Differentiable Graph Partitioning for Protein Representation Learning
1. This paper introduces BIOBLOBS, a novel module for protein representation learning that dynamically partitions proteins into flexible, non-overlapping substructures, termed “blobs”. This approach captures the modular nature of protein function more effectively than traditional methods relying on rigid substructures.
2. BIOBLOBS is fully differentiable and can be integrated into existing protein encoders like GVP-GNN. It uses a neural partitioning layer to create these blobs, which are then quantized into a shared codebook, resulting in discrete, interpretable representations of protein substructures that improve performance on various protein function prediction tasks.
3. The neural partitioner in BIOBLOBS employs a seed-and-expand strategy to form cohesive substructures. It selects seed residues using Gumbel-Softmax sampling and expands around them based on learned thresholds, ensuring that the resulting blobs are connected and functionally relevant.
4. A key innovation is the use of a vector-quantization codebook to map blob embeddings to discrete tokens. This not only captures frequent and functionally relevant substructures across the dataset but also enables the model to learn a compact and reusable vocabulary of protein motifs.
5. The global-blob attention fusion module integrates the quantized blob embeddings with the global protein representation. This allows the model to attend to informative substructures and produces an interpretable importance score distribution over blobs, enhancing the model’s interpretability.
6. Experiments across three protein function prediction benchmarks (Gene Ontology, Enzyme Commission, and Structural Class) demonstrate that BIOBLOBS outperforms strong baselines under both random and structure-based splits. The improvements are particularly significant on the structure split, which reduces similarity leakage.
7. The authors provide a detailed analysis of the model’s partitions, showing that BIOBLOBS consistently identifies well-defined secondary structures and assigns high importance scores to coherent, stable structures. The codebook further maps similar substructures to nearby codes, providing a meaningful vocabulary.
8. BIOBLOBS addresses the challenge of selecting substructures of variable size and topology by introducing a differentiable seed-and-expand procedure. This, coupled with discrete codebook learning, yields more faithful protein representations and a scalable account of structure-function relationships.
💻Code: github.com/OliverLaboratory/…
📜Paper: arxiv.org/abs/2510.01632
#ProteinRepresentationLearning #GraphPartitioning #NeuralPartitioner #ProteinFunctionPrediction #DeepLearning #ComputationalBiology
1
3
23
1,687
20 Sep 2025
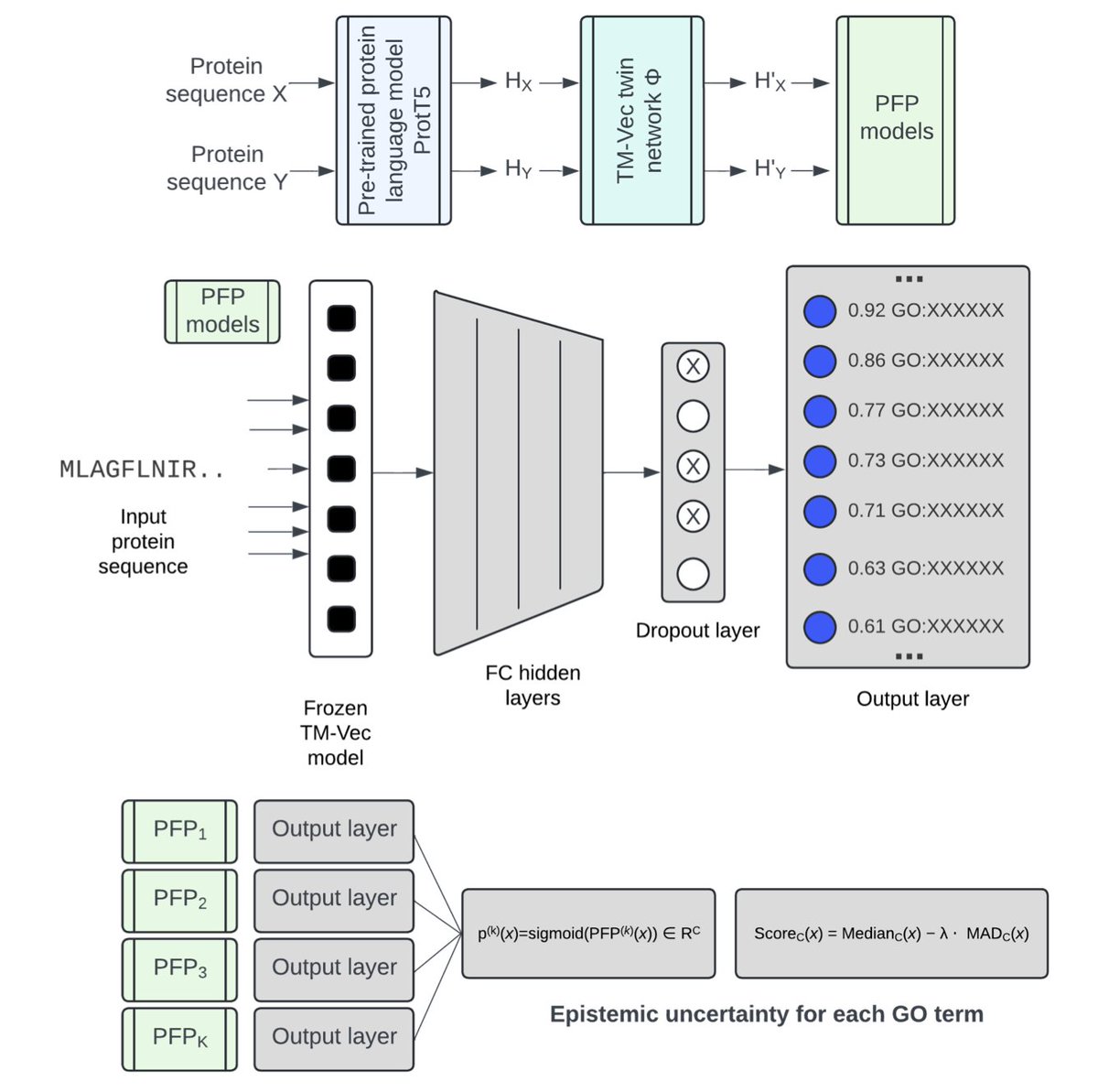
PlasmoFP: leveraging deep learning to predict protein function of uncharacterized proteins across the malaria parasite genus
1. A new deep learning model, PlasmoFP, has been developed to predict the functions of uncharacterized proteins in the malaria parasite genus Plasmodium. This is a significant step forward in understanding the biology of these parasites, which cause millions of infections and thousands of deaths each year.
2. PlasmoFP models are trained on structure-function relationships of proteins from the SAR supergroup, which includes Plasmodium. This approach addresses the challenge of low sequence similarity between Plasmodium proteins and well-characterized proteins from other organisms.
3. The models estimate epistemic uncertainty and control false discovery rates, outperforming existing methods. By integrating PlasmoFP predictions with existing annotations, the proportion of fully annotated proteins across Plasmodium species increased significantly, improving proteome-wide annotation completeness.
4. PlasmoFP predictions help advance Plasmodium basic research, which can aid progress towards global malaria control. The model's ability to predict functions for partially annotated and 'unknown function' proteins is a critical resource for developing novel therapeutic and diagnostic strategies.
5. The study demonstrates that PlasmoFP can effectively predict meaningful GO terms for partially annotated proteins, extending its benefits beyond proteins of unknown function. This is particularly important for less-studied Plasmodium species, where annotation coverage is limited.
6. PlasmoFP models maintain high performance even on proteins with regions of intrinsic disorder, which are challenging for traditional sequence-based methods. This suggests that the models can learn relevant structure-function features from disordered proteins.
7. The researchers used a unique approach to quantify uncertainty in model predictions, employing deep ensembles to improve model precision and reduce false discovery rates. This method provides a more reliable prediction set with explicit error-rate control.
📜Paper: biorxiv.org/content/10.1101/…
#DeepLearning #ProteinFunctionPrediction #Malaria #Plasmodium #Bioinformatics #Genomics
3
1,108
13 Sep 2025
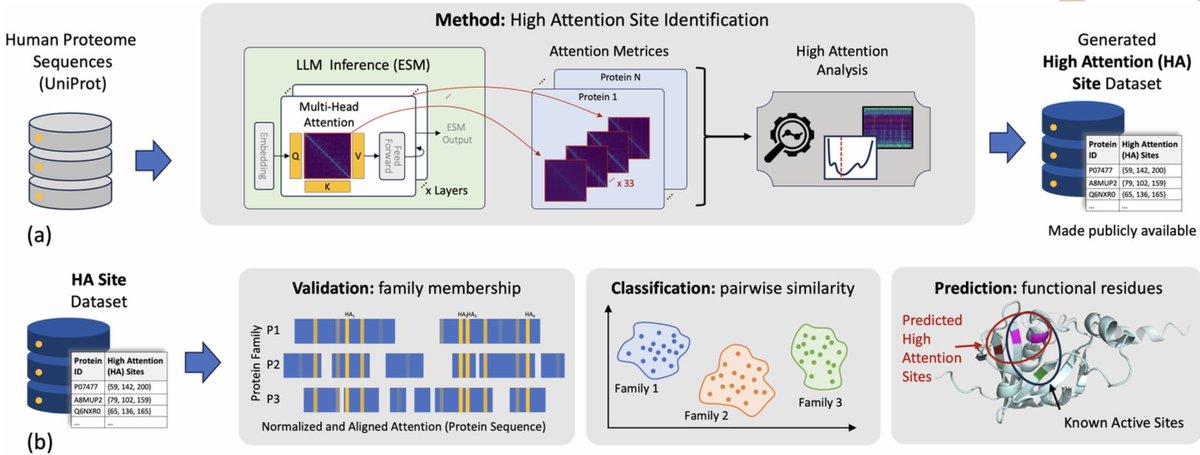
Paying attention to attention: High attention sites as indicators of protein family and function in language models @PLOSCompBiol
1. A novel approach has been introduced to identify High Attention (HA) sites within protein primary sequences using the Evolutionary Scale Modelling (ESM) language model. These HA sites correspond to key residues that define protein families and functions.
2. The study demonstrates that HA sites provide interpretable links to biological functions. By examining attention patterns across multiple layers of the ESM model, researchers pinpoint residues that contribute most to family classification and function prediction.
3. HA sites improve active site predictions for functions of unannotated proteins. This suggests that protein language models implicitly capture functional signals within protein sequences, enhancing the interpretability of these models.
4. The research makes available the HA sites for the human proteome, offering a broadly applicable approach to protein classification and functional annotation. This resource can facilitate studying key sites in each protein.
5. The method systematically identifies key residues in the middle layers of the protein language model, where attention converges on these HA sites. This provides a clearer understanding of how the model distinguishes between different protein families.
6. The study shows that the HA sites often overlap with biologically important residues, such as active sites. This highlights the model's ability to focus on functionally relevant regions of the protein.
7. The HA sites are not solely governed by regions of high sequence conservation. They can be found at positions with low consensus and still be functionally relevant, indicating that the model captures more than just simple sequence similarity.
8. The research also evaluates the correlation between HA sites and protein structure, finding that while HA sites are identified early in the model's attention layers, they do not necessarily correlate with structural importance, suggesting a distinction between functional and structural signals in the model.
9. The study concludes that HA sites are early detectors of protein function and can be used to define protein families. They are robust across proteins with varying sequence lengths and are consistent across members of the same protein family.
10. The findings suggest that protein language models first identify broad family-specific features before refining more detailed representations in deeper layers, with HA sites playing a key role in this initial categorization.
📜Paper: journals.plos.org/ploscompbi…
#ProteinLanguageModels #HighAttentionSites #ProteinFunctionPrediction #ComputationalBiology #Bioinformatics
14
51
3,828
12 Aug 2025
Protein Function Prediction via Contig-Aware Multi-Level Feature Integration
1. A novel study introduces CAML, a novel deep learning model for protein function prediction that integrates both intra-protein features (sequence and structure) and inter-protein features (CDS topological relationships within contigs). This approach significantly outperforms existing methods in accuracy, precision, recall, and F1-score.
2. CAML leverages a Graph Isomorphism Network (GIN) to extract structural features from predicted protein contact graphs and uses ESM-2 for sequence embeddings. It also employs a BiLSTM to model functional relationships among colocalized CDSs within contigs, capturing operon-like associations. This multi-level feature integration is critical for enhancing functional annotation accuracy.
3. The study demonstrates that incorporating contig topology is essential for accurate protein function prediction. CAML achieves improvements of up to 56.64% in accuracy and 60.84% in F1-score over the second-best model. This highlights the importance of considering both protein structure and genomic context for robust functional annotation.
4. CAML is database-independent and can generalize across different annotation systems, such as from COG to GO annotations. This flexibility makes it a powerful tool for genomics research, bridging the gap between genomic context and protein function.
5. The model's architecture includes four main modules: protein graph construction, protein-level feature extraction, protein feature enhancement, and contig-level topology-aware feature extraction. This modular design allows for a comprehensive integration of sequence, structure, and genomic context.
6. Experiments on a benchmark dataset with 15,000 contigs demonstrate CAML's superior performance across all evaluation metrics. The model shows remarkable robustness, especially in handling imbalanced datasets with varying protein abundance levels.
7. Ablation studies confirm the critical contributions of ESM-2 embeddings, GIN-based feature aggregation, and contig topological information. These components are essential for capturing the intricate relationships between protein structure, sequence, and genomic architecture.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinFunctionPrediction #DeepLearning #ComputationalGenomics #Bioinformatics #ContigTopology #CAML

4
23
1,649
10 Aug 2025
AstraROLE2 & AstraSUIT2: Multi-Task Annotation Models for Functional Profiling of Proteins
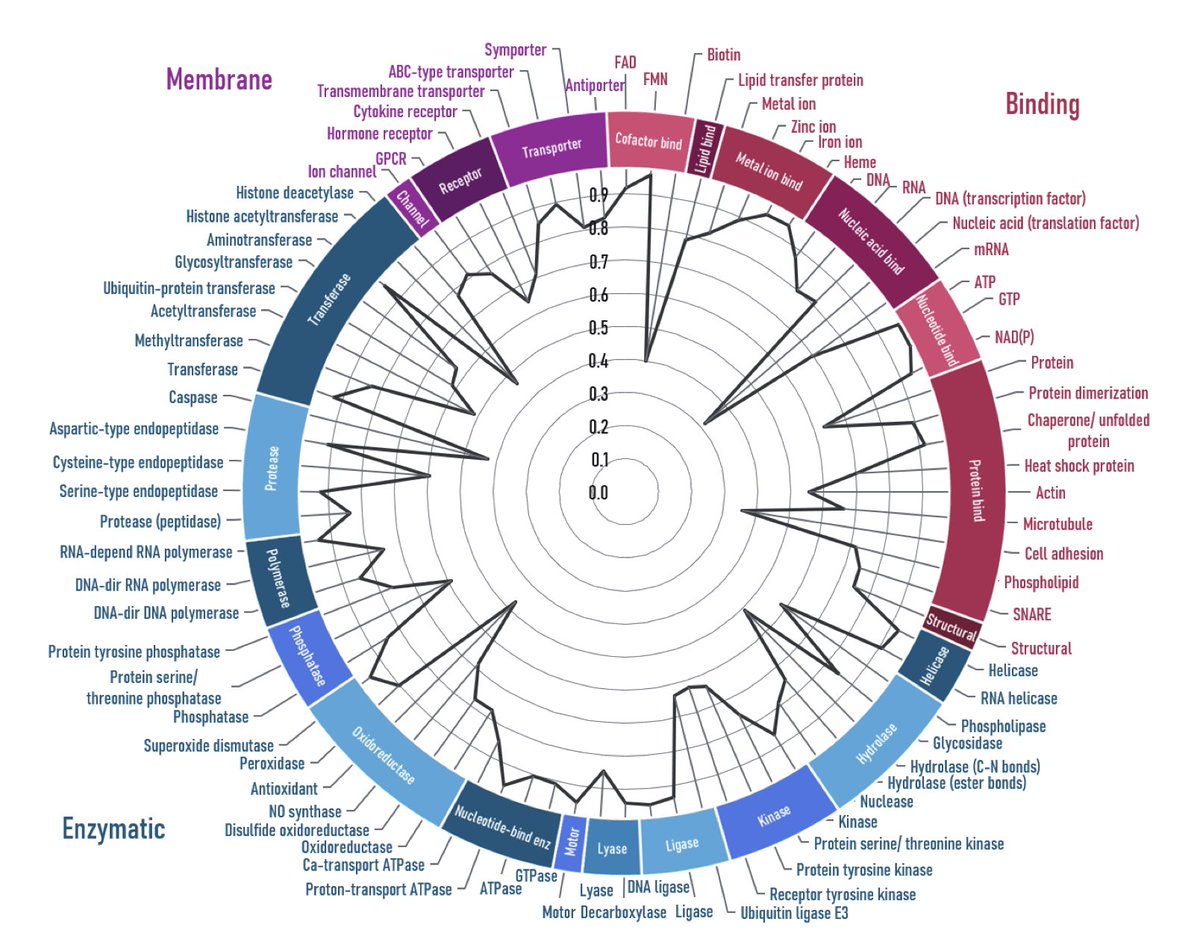
1. A new study introduces AstraROLE2 and AstraSUIT2, two transformer-based multi-task models that provide comprehensive functional profiles of proteins in a single unified framework. These models address the limitation of existing tools that focus on only one aspect of protein function by integrating multiple annotations into a single model.
2. The models use a 1,351-dimensional input vector, combining ESM-2CLS embeddings and physicochemical features. They are trained on over 730,000 UniProt proteins, with stratified splits and class-weighted BCE loss to handle imbalances. AstraROLE2 predicts EC class, GO terms, molecular pathways, and protein categories, while AstraSUIT2 annotates cofactor binding, domains, membrane associations, and more.
3. AstraROLE2 and AstraSUIT2 achieve macro F1 scores of 0.84–0.98 and MCC scores of 0.85–0.98 on hold-out sets. The models excel in predicting cofactor binding (F1=0.98) and membrane association types (F1=0.97), outperforming recent comparators like DeepGOPlus and TargetP 2.0 in metal-ion and cofactor binding tasks.
4. Case studies on three novel proteins (IgtG, esmGFP, and TPM3P9) demonstrate the models' ability to generate accurate predictions for most labels, even for proteins with high novelty. This highlights their potential for hypothesis generation and validation in experimental research.
5. The study concludes that AstraROLE2 and AstraSUIT2 offer state-of-the-art performance in multi-label protein annotation, providing a valuable tool for researchers. Future work will focus on incorporating more training data and exploring synergies between different models to further enhance prediction capabilities.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinAnnotation #AIinBiology #ComputationalBiology #MultiTaskLearning #ProteinFunctionPrediction
1
7
1,331
3 Aug 2025
SuperEdgeGO: Edge-supervised graph representation learning for enhanced protein function prediction @PLOSCompBiol
1. A new computational method called SuperEdgeGO has been proposed to predict protein functions more accurately. This method leverages edge-supervised graph representation learning to enhance the prediction of protein functions, addressing the limitation of previous methods that underutilized edge information in protein graphs.
2. SuperEdgeGO introduces a supervised attention mechanism to explicitly encode residue contacts into protein representations. Unlike traditional graph convolution methods that use edge information in an unsupervised manner, this approach directly supervises the edges, leading to more effective capture of structural features.
3. The study demonstrates that SuperEdgeGO achieves state-of-the-art performance across all three categories of protein functions (molecular function, biological process, and cellular component). The ablation analysis further validates the effectiveness of the edge supervision strategy.
4. SuperEdgeGO uses AlphaFold2-predicted protein structures to construct protein graphs, which are then processed through a novel attention mechanism. This method not only improves the accuracy of function prediction but also highlights the importance of edge information in protein structure modeling.
5. The model's performance is evaluated on a benchmark dataset containing over 20,000 human proteins. SuperEdgeGO outperforms existing state-of-the-art methods, particularly in the molecular function category, where it achieves a significant improvement in Fmax.
6. The authors also conducted cross-species experiments on datasets from different organisms, including S. cerevisiae, E. coli, fruit fly, and rat. The results show that SuperEdgeGO can generalize well across species, further proving its effectiveness.
7. The edge supervision strategy in SuperEdgeGO has the potential to be applied to other biological tasks that rely on structural insights, such as drug-target affinity prediction and protein-protein interaction analysis.
💻Code: github.com/Lyt0715/SuperEdge…
📜Paper: journals.plos.org/ploscompbi…
#ProteinFunctionPrediction #GraphRepresentationLearning #ComputationalBiology #SuperEdgeGO #EdgeSupervision #AlphaFold2

3
26
1,803
31 Jul 2025
Scaling and Data Saturation in Protein Language Models
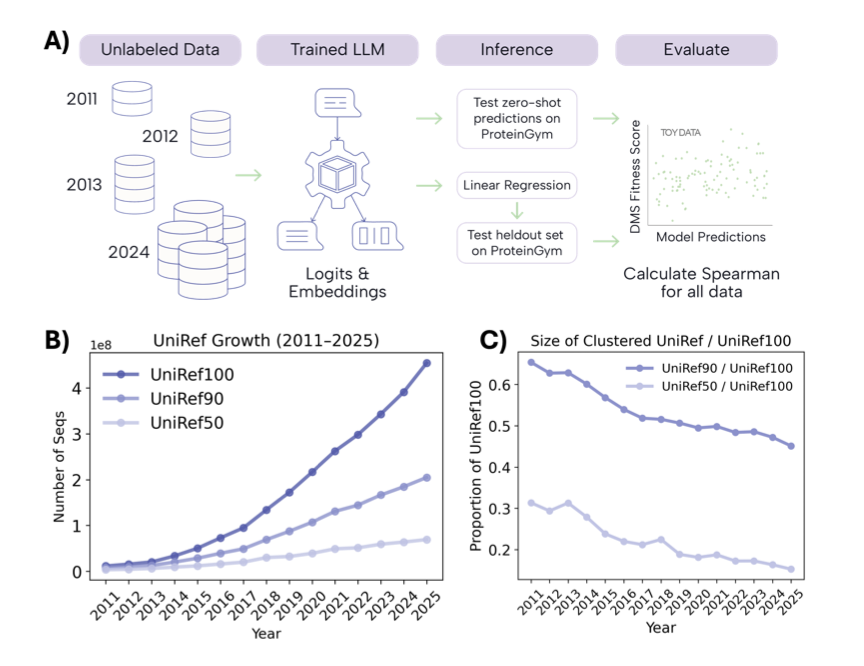
1. This study investigates how protein language models (pLMs) scale with increasing pretraining data, focusing on protein function prediction. The research uses yearly snapshots of UniRef100 from 2011 to 2024 and evaluates model performance using the ProteinGym benchmark.
2. The study finds that model performance does not improve monotonically with more data. Instead, performance fluctuates year-to-year, indicating that protein language models have not yet reached data saturation for robust generalization.
3. The research highlights the importance of targeted data acquisition. While large-scale pretraining data is essential, the specific composition of the data significantly impacts model performance. This underscores the need for strategic data curation.
4. The study also explores semi-supervised learning using ridge regression on sequence embeddings. It shows that even a small amount of labeled data can significantly boost performance, highlighting the value of experimental labels.
5. The authors use the well-characterized β-Lactamase protein to test if scaling laws emerge with abundant data. They find that unsupervised predictions improve over time, and semi-supervised learning can match the performance of larger models with just one experimental dataset.
6. The study concludes that biological data scaling for protein function prediction is complex and does not follow a simple trend. Continued exploration of data scaling across different families, tasks, and learning paradigms is needed to guide the development of future biological language models.
7. The authors provide all training, inference, analysis, and visualization code on GitHub, allowing the community to reproduce and build upon their work. This transparency is crucial for advancing research in protein language modeling.
📜Paper: arxiv.org/abs/2507.22210
#ProteinLanguageModels #DataScaling #Bioinformatics #MachineLearning #ProteinFunctionPrediction
7
902
24 Jul 2025
Opus-go: Unlocking Residue-Level Insights from Sequence-Level Annotations Using Biological Language Models
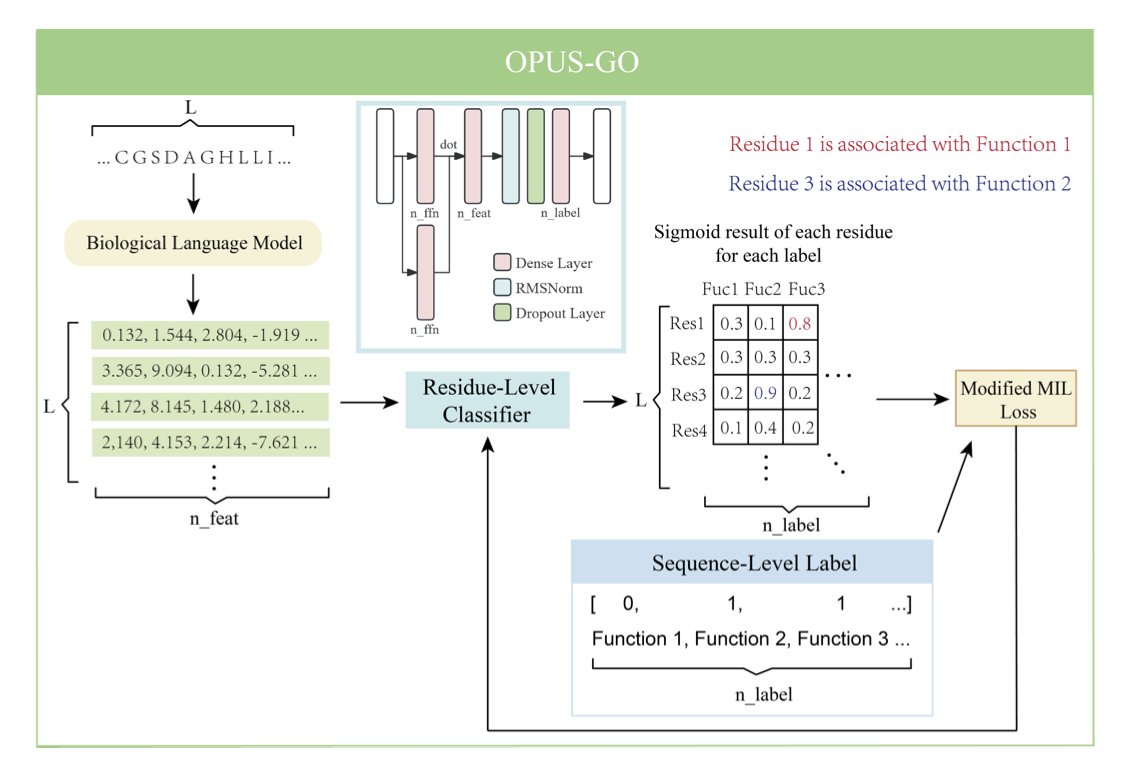
1. OPUS-GO is a novel method that enhances protein function prediction by providing both sequence-level accuracy and residue-level interpretability. This approach is particularly innovative as it bridges the gap between high-level classification and detailed residue analysis, offering deeper biological insights.
2. The core of OPUS-GO lies in its modified Multiple Instance Learning (MIL) strategy. By treating protein sequences as bags and residues as instances, it effectively identifies critical residues associated with functional labels. This dual-level approach outperforms traditional methods in both sequence and residue classification tasks.
3. OPUS-GO leverages biological language models (BLMs) like ESM-2 to extract features, ensuring that the method is broadly applicable and can be integrated into various existing frameworks. Its ability to pinpoint critical residues without requiring structural information makes it a powerful tool for functional annotation.
4. In experiments across multiple benchmarks, OPUS-GO consistently demonstrates superior performance. For example, it achieves higher AUPR and Fmax scores in Gene Ontology (GO) term prediction tasks compared to baseline methods. This highlights its robustness and accuracy in identifying functional residues.
5. The method also shows promise in enzyme function prediction, identifying consistent patterns within enzyme categories. While it may not always pinpoint specific active sites, its ability to detect key residues related to functional labels makes it a valuable asset for protein design and engineering.
6. The code and pre-trained models for OPUS-GO are publicly available, allowing researchers to easily implement and build upon this work. This accessibility is crucial for advancing the field and encouraging further research into protein function prediction and design.
💻Code: github.com/thuxugang/opus_go
📜Paper: openreview.net/forum?id=iYXa…
#ProteinFunctionPrediction #Bioinformatics #MachineLearning #ComputationalBiology
2
820
24 Jul 2025
Opus-go: Unlocking Residue-Level Insights from Sequence-Level Annotations Using Biological Language Models
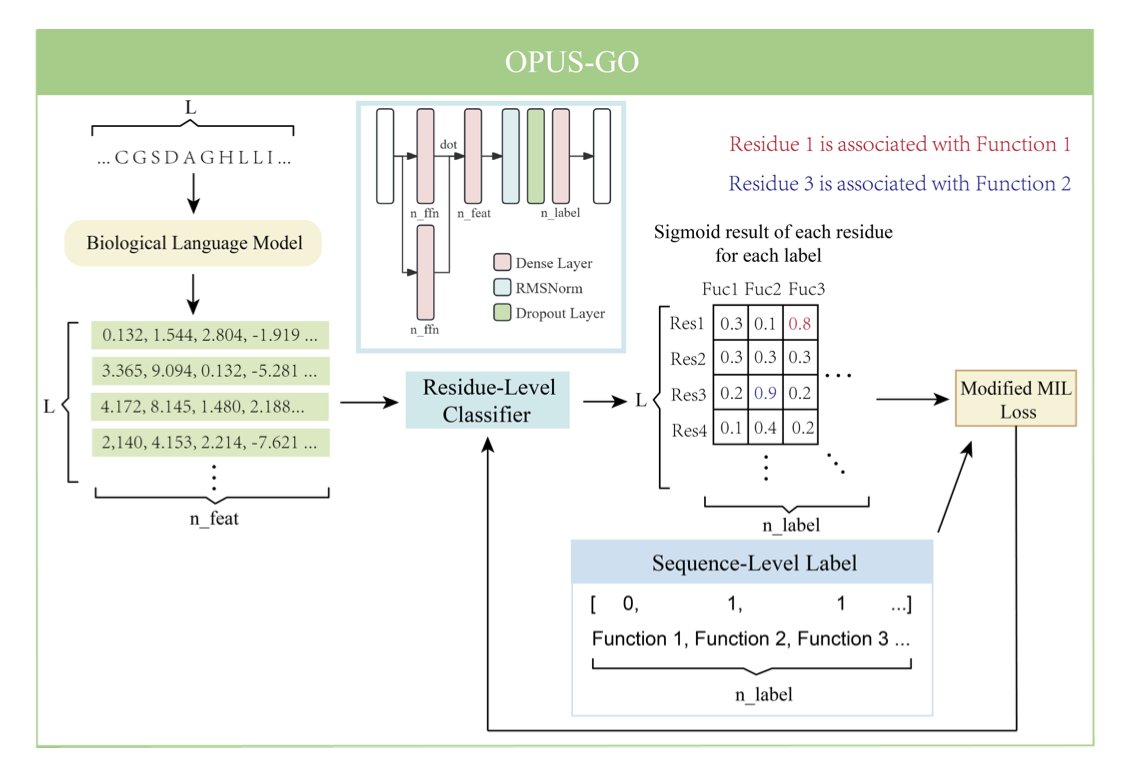
1. OPUS-GO is a novel method that enhances protein function prediction by providing both sequence-level accuracy and residue-level interpretability. This approach is particularly innovative as it bridges the gap between high-level classification and detailed residue analysis, offering deeper biological insights.
2. The core of OPUS-GO lies in its modified Multiple Instance Learning (MIL) strategy. By treating protein sequences as bags and residues as instances, it effectively identifies critical residues associated with functional labels. This dual-level approach outperforms traditional methods in both sequence and residue classification tasks.
3. OPUS-GO leverages biological language models (BLMs) like ESM-2 to extract features, ensuring that the method is broadly applicable and can be integrated into various existing frameworks. Its ability to pinpoint critical residues without requiring structural information makes it a powerful tool for functional annotation.
4. In experiments across multiple benchmarks, OPUS-GO consistently demonstrates superior performance. For example, it achieves higher AUPR and Fmax scores in Gene Ontology (GO) term prediction tasks compared to baseline methods. This highlights its robustness and accuracy in identifying functional residues.
5. The method also shows promise in enzyme function prediction, identifying consistent patterns within enzyme categories. While it may not always pinpoint specific active sites, its ability to detect key residues related to functional labels makes it a valuable asset for protein design and engineering.
6. The code and pre-trained models for OPUS-GO are publicly available, allowing researchers to easily implement and build upon this work. This accessibility is crucial for advancing the field and encouraging further research into protein function prediction and design.
💻Code: github.com/thuxugang/opus_go
📜Paper: openreview.net/forum?id=iYXa…
#ProteinFunctionPrediction #Bioinformatics #MachineLearning #ComputationalBiology
1
2
820
27 Mar 2025
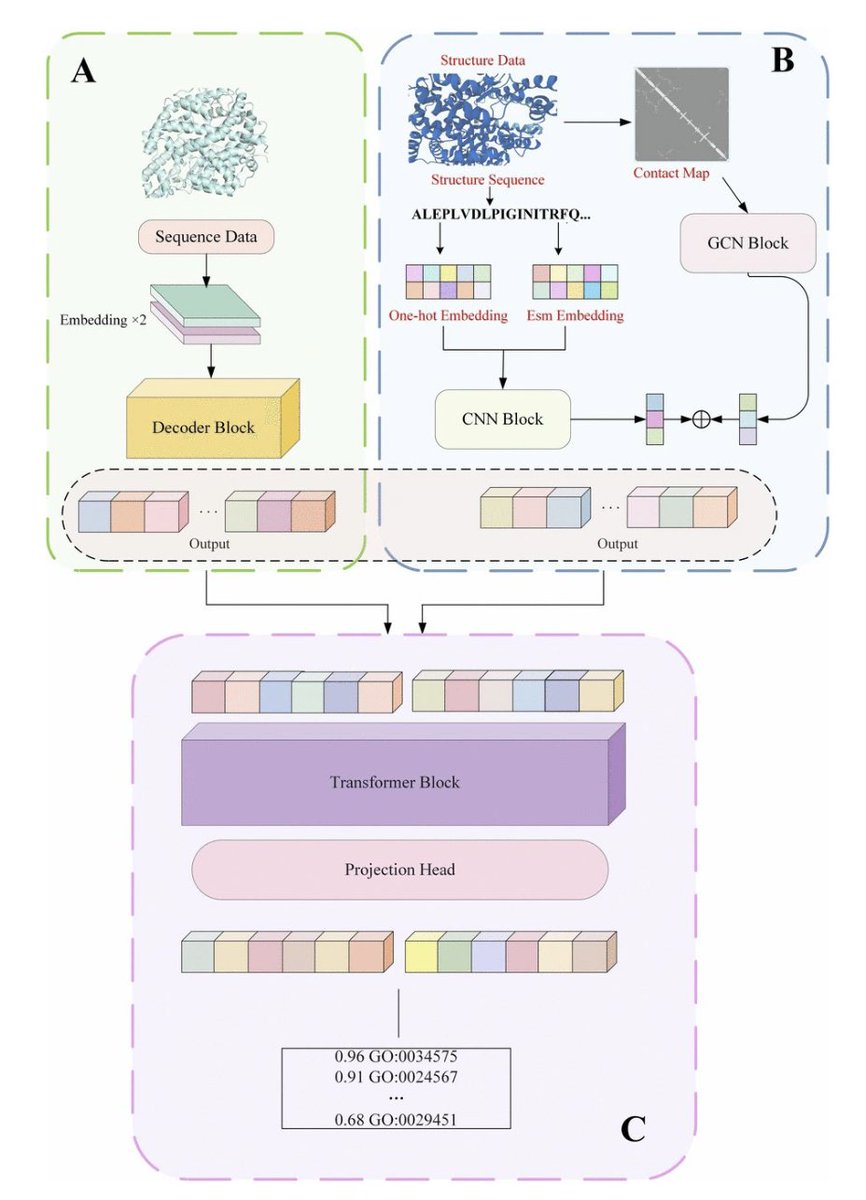
A Multimodal Model for Protein Function Prediction (MMPFP) @SciReports
1. MMPFP is a novel multimodal model for protein function prediction that integrates both protein sequence and structure information using Graph Convolutional Networks (GCN), Convolutional Neural Networks (CNN), and Transformer models. This approach addresses the limitations of single-modality models that often overlook structural properties critical for accurate predictions.
2. The model processes inputs through three main modules: the protein sequence encoding module, the multilayer GCN protein representation module, and the protein CNN module. By combining these modules, MMPFP constructs a comprehensive framework for learning complex protein functions.
3. MMPFP achieves state-of-the-art performance in predicting Molecular Function (MF), Biological Process (BP), and Cellular Component (CC) with AUPR scores of 0.721, 0.401, and 0.495, respectively. Fmax scores of 0.769, 0.632, and 0.695, and Smin scores of 0.320, 0.480, and 0.448 demonstrate significant improvements over baseline models.
4. Ablation studies confirm that the Transformer module within the GCN branch is essential for capturing complex relationships within protein graphs, providing a substantial performance boost over LSTM-based methods.
5. The combination of CNN, GCN, and Transformer modules allows the model to effectively integrate spatial structural features and sequence information, enhancing the overall prediction accuracy and robustness.
6. Comparative analysis against various baseline models, including TAWFN, DeepGO, DeepFRI, and others, shows that MMPFP consistently outperforms these methods by 3-5% in Fmax, AUPR, and Smin metrics.
7. The model’s multimodal architecture allows for a more comprehensive understanding of protein function, making it a promising tool for tasks like protein structure prediction, multitask learning, and integrating additional modalities such as protein-protein interaction networks.
8. Future work aims to expand MMPFP’s capabilities by incorporating new learnable features and advanced deep learning models to further improve prediction accuracy and broaden its applicability.
📜Paper: nature.com/articles/s41598-0…
#ProteinFunctionPrediction #MultimodalLearning #GCN #CNN #Transformer #DeepLearning #Bioinformatics #ProteinStructure
577