
Harbour BioMed sets up Megastream TechBio with Baidu-barked Biomap to combine data with LLM models to develop innovative biologics. allsci.com/news/licensing-de…
46
feverfew retweeted

🇨🇳Harbour BioMed and 🇨🇳BioMap will launch MegaStream TechBio, a global pipeline company integrating exclusive datasets, purpose‑built large models, and a large‑scale innovative portfolio into a unified AI‑powered R&D engine. harbourbiomed.com/news/271.h…
MegaStream will combine a multimodal generative large model, an intelligent dry–wet closed‑loop discovery lab, and a broad FIC/BIC pipeline spanning cardiovascular, renal, oncology and anti‑aging diseases.
The company aims to advance differentiated complex biologics into the clinic at scale and emerge as a leading AI‑native biologics developer.
Harbour BioMed will contribute its Harbour Mice® fully human antibody platform, deep target biology expertise, and global development capabilities.
BioMap brings its xTrimo life‑science foundation models, model engineering, and automated discovery infrastructure.
MegaStream’s initial pipeline includes AI‑identified candidates from prior collaborations plus new AI‑driven programs. Founding partners will receive potential upfronts, milestones and royalties.
The alliance is built on three pillars:
1⃣AI‑driven AIDD infrastructure optimized for complex biologics, including multispecifics, XDCs, in vivo CAR‑T and inhaled/oral biologics.
2⃣A next‑generation dry–wet closed‑loop lab enabling automated DBTL cycles, >500% efficiency gains, and >5 PB of AI‑ready data over five years.
3⃣Scaled FIC/BIC pipelines, leveraging generative AI to replace traditional screening with predictive design and targeted validation.
1
7
902

Jun 16
🧬 NUEVO INFORME DE INVERSIÓN: $RXRX (Recursion Pharmaceuticals)
¿Es una biotech tradicional o una plataforma de software con opcionalidad asimétrica? Analizo a fondo su modelo "TechBio", el respaldo de NVIDIA y sus catalizadores clínicos.
Lee mi informe completo aquí abajo 👇

2
2
24
6,703

Jun 16
Sequence-based drug-target interaction (DTI) frameworks are proving a critical point: you don't always need complex 3D protein structures to achieve accurate virtual screening.
But scaling these individual deep learning pipelines across an entire R&D pipeline is where enterprise teams run into a bottleneck.
That’s exactly why we built the Boltzmann AI Discovery Suite. Our autonomous multi-agent platform unifies sequence-based machine learning, molecular docking, and property prediction into a single, intuitive workspace.
We help biotech and pharma teams move from raw genomic data to synthesizable leads in days instead of months.
🔬 Accelerate your pipeline: Explore our autonomous research workflows at boltzmann.co/
#DrugDiscovery #TechBio #GenerativeAI #Bioinformatics #PharmaTech
Jun 15

Sequence-based prediction of drug–target binding using machine learning, deep learning and ensemble models without 3D structural information
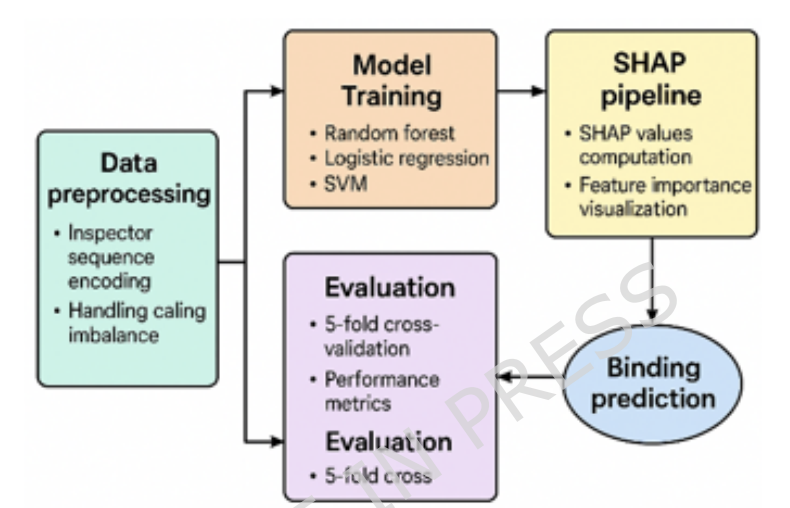
1. The study presents a fully sequence-based drug–target interaction (DTI) pipeline designed to work when reliable 3D protein structures are missing, aiming for docking-comparable discrimination while keeping the feature space interpretable.
2. Core idea: a unified, hand-engineered representation that concatenates (i) protein physicochemical descriptors (e.g., hydrophobicity/charge/composition), (ii) BLOSUM62-derived evolutionary information, (iii) protein 3-gram motif frequencies (local sequence context), and (iv) sequence-like drug encodings derived from canonical SMILES motif frequencies.
3. The framework is intentionally model-agnostic: it evaluates classical ML (Logistic Regression, SVM, Random Forest), deep learning (MLP, CNN), and multiple ensemble approaches (Extra Trees, Gradient Boosting, Histogram-based Gradient Boosting), plus a stacking ensemble.
4. The stacking classifier combines Random Forest SVM Logistic Regression via a meta-learner, leveraging complementary decision boundaries; reported mean ROC-AUC exceeds 0.90, with a maximum AUC of 0.914 under the paper’s protocol.
5. Evaluation emphasizes methodological hygiene: stratified 5-fold cross-validation, with fold-wise preprocessing to reduce leakage risk; scaling and SMOTE are applied only within training folds (validation/test folds remain untouched).
6. SMOTE’s impact is analyzed explicitly and shown to be model-dependent: it often improves minority-class sensitivity/recall, while effects on Accuracy/F1/ROC-AUC can vary across architectures—highlighting why imbalance handling must be reported alongside metrics.
7. Beyond scalar metrics, the paper inspects learning dynamics for deep models (CNN convergence behavior), confusion matrices for representative models, and ROC curves to characterize threshold-independent discrimination across folds.
8. Interpretability is treated as a first-class goal: permutation importance and SHAP analyses identify influential features, with protein-derived features (physicochemical properties and specific 3-gram motifs) frequently dominating—supporting biologically grounded explanations rather than opaque latent embeddings.
9. For orthogonal validation, the authors perform molecular docking (AutoDock Vina) on selected predicted pairs, using PDB structures and/or AlphaFold2 models filtered by confidence (pLDDT/PAE). A showcased case (EGFR) aligns high predicted binding probability with favorable docking scores (e.g., around −6.4/−6.2 kcal/mol), used as qualitative support.
10. Limitations are acknowledged: no independent external test set; stratified CV may still be optimistic if similar proteins/ligands appear across folds; docking validation is illustrative rather than a dataset-wide quantitative correlation—future work proposed includes stricter splits (cold-start/sequence-identity/scaffold splits) and broader docking benchmarks.
📜Paper: doi.org/10.1038/s41598-026-5…
#DrugDiscovery #ComputationalBiology #Bioinformatics #MachineLearning #DeepLearning #EnsembleLearning #DTI #Cheminformatics #ExplainableAI #VirtualScreening
3
39

🤖🧬 New AI-Powered TechBio Platform Unveiled
@Harbour_BioMed and BioMap have launched the MegaStream AI-powered TechBio platform.
👉 cgxpwire.com/technology/harb…
#AI #TechBio #DrugDiscovery #Innovation

4

Jun 15
Harbour BioMed and BioMap Jointly Initiate MegaStream TechBio: A Leading Global Drug ... prnewswire.com/news-releases… #GenerativeAI #GenAI #AI
9

Jun 15
Harbour BioMed and BioMap Jointly Initiate MegaStream TechBio: A Leading Global Drug Development Platform Joins Forces with Premier Life Science Foundation Models to Set New Benchmarks for AI-Driven Complex Biologics R&D dlvr.it/TT31kZ
14
Anne Marie Droste retweeted

Jun 13
one slight issue I often have with techbio is the way people use some very specific model benchmark as validation that their tool is somehow “better”. i think in general we need a better push for more top down biologically informed benchmarks that truly ask “will this make my drug better at what it needs to do” especially in cancer vaccines
1
1
19
8,324

Jun 12
from my pov, the primary objective for the block does indeed seem to be safety, but there is a secondary effect of deterring techbio companies from making progress, which incidentally aligns with anthropic's increasing focus on biology
16

It is still too early start a techbio!!

44
antisense. retweeted

TechBio Companies per category Mapping. Source: MMC

7
16
91
18,972

Jun 12
TechBio talent tenacity are powering Switzerland’s next wave of innovation. 🧬🤖
From dual education and global talent to academia-industry collaboration and deeptech patience, the Swiss edge is built to last. Read more in the Swiss Biotech Report 2026: swissbiotech.org/news/biotec…

2
32

Jun 11
Proteins are the machines our bodies run on, and predicting how they interact is central to designing new treatments. In the wake of AlphaFold’s 2024 Nobel win, what’s the state of AI models for proteins, and how do techbio business models impact which scientists can use them?
I recently sat down with @GabriCorso, CEO and co-founder of @boltz_bio, a venture-backed startup driven by opening up scientist access to these models.
In our conversation we covered:
The shift from discovering drugs by chance to designing them with AI models (Boltz-1, Boltz-2, and BoltzGen)
Boltz's focus on openness, and how it diverges from fully integrated efforts like DeepMind's Isomorphic Labs
Boltz’s multi-year partnership with @pfizer, and how proprietary data sharpens models for a company's specific drug-discovery efforts
The road to "zero-shot drugs," autonomous labs, and the future of bio R&D
If you're curious about where frontier AI meets drug discovery, check out this latest episode of the Discovery Engines Podcast.
👀 YouTube: youtu.be/fLY5yhRNEKg
🎧 Spotify: open.spotify.com/episode/50K…
5
27
7,777