
Apr 16
📢 #highlycited paper
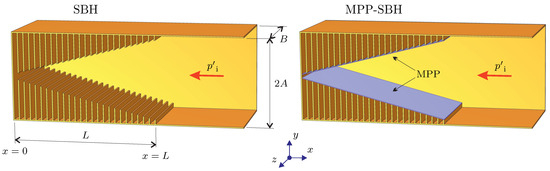
📚 Maximizing the #AbsorbingPerformance of Rectangular #SonicBlackHoles †
🔗 mdpi.com/2076-3417/14/17/776…
👨🔬 by Milan Červenka et al.
🏫 Czech Technical University in Prague
#rectangulargeometry #evolutionaryoptimization
1
2
30

29 Jul 2025
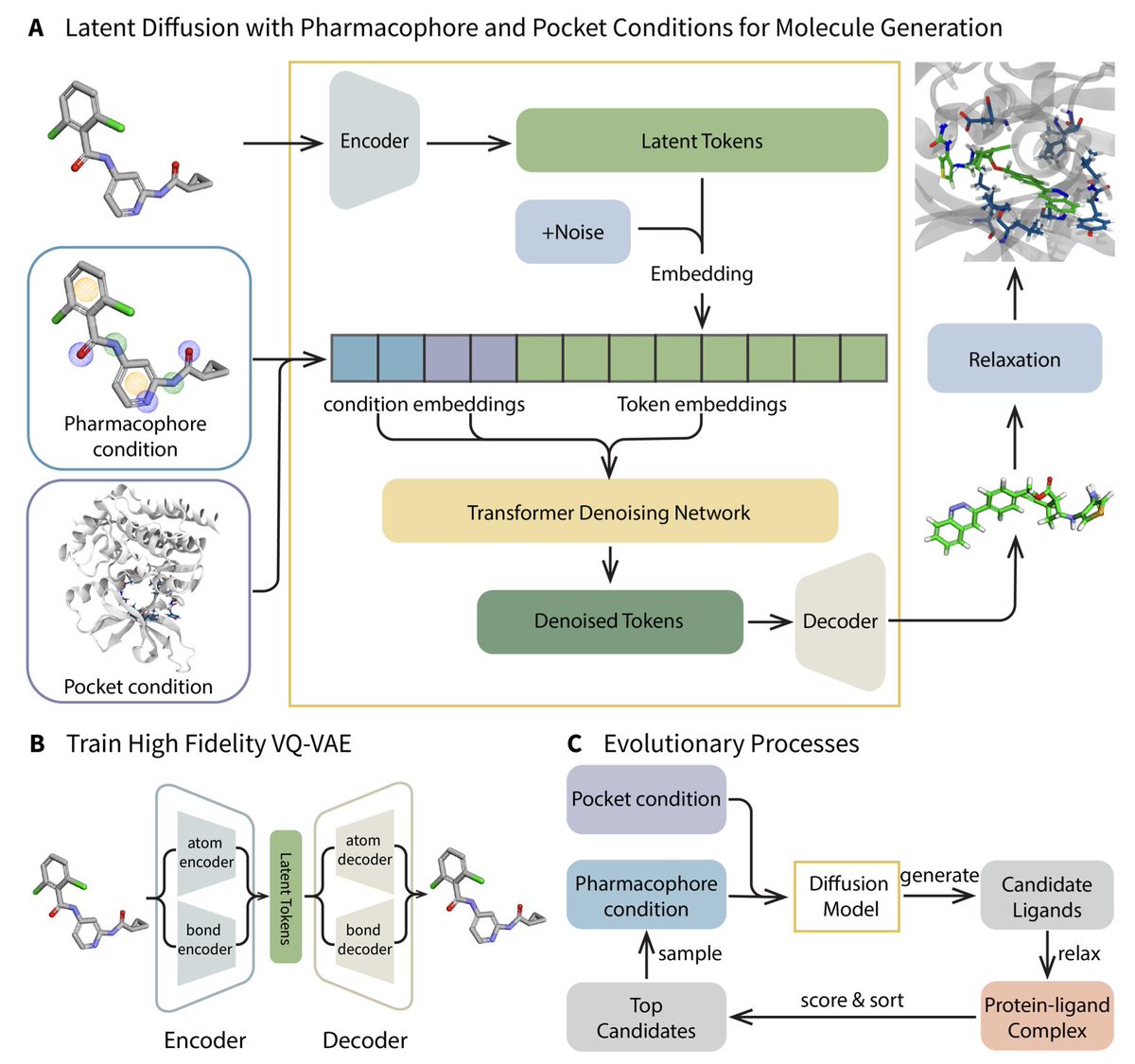
Generative Molecule Evolution Using 3D Pharmacophore for Efficient Structure-Based Drug Design
1. A groundbreaking framework named MEVO has been introduced to bridge the gap between large-scale small molecule datasets and limited protein-ligand complex datasets, significantly enhancing the training data for structure-based drug design (SBDD) models. This innovative approach leverages pharmacophore-guided molecule generation and evolutionary optimization strategies to design high-affinity binders for various protein targets.
2. MEVO incorporates three key components: a high-fidelity VQ-VAE for molecule representation in latent space, a diffusion model for pharmacophore-guided molecule generation, and a pocket-aware evolutionary strategy for optimizing molecules with physics-based scoring functions. This combination allows for the efficient generation of molecules that fit both pharmacophore principles and target-binding constraints.
3. The framework demonstrates remarkable versatility and generalizability, validated through the design of potent inhibitors for KRASG12D, a challenging cancer target. MEVO-generated molecules exhibit binding affinities comparable to known highly active inhibitors, as evaluated by free energy perturbation (FEP) calculations.
4. MEVO effectively addresses the limitations of current SBDD methods by integrating extensive training data from ligand-only datasets, which are almost unlimited compared to protein-ligand complex datasets. This strategy allows the model to learn robust pharmacophore patterns and generate diverse and plausible molecules.
5. The evolutionary optimization strategy in MEVO uses a physics-informed scoring function to iteratively improve the binding affinity of generated molecules. This training-free approach can be easily transferred to other tasks, such as optimizing ligand properties like permeability and solubility.
6. MEVO generates drug-like molecules with desirable properties, as evidenced by comparisons with FDA-approved drugs. The generated molecules comply with Lipinski’s rule of five, exhibit reasonable drug-likeness scores, and have similar logP distributions to reference molecules.
7. In hit discovery, MEVO successfully identifies high-affinity binders for various protein targets, outperforming known binders in some cases. The model’s ability to preserve critical interactions within binding pockets provides a strong foundation for developing potent ligands.
8. MEVO’s high versatility is showcased through its application in different stages of drug design, including ligand growing, linker design, and lead optimization. The model’s ability to design high-affinity binders for KRASG12D highlights its potential in tackling challenging drug targets.
📜Paper: arxiv.org/abs/2507.20130
#DrugDesign #GenerativeModels #StructureBasedDrugDesign #Pharmacophore #EvolutionaryOptimization #KRASInhibitors #AIinPharma
4
8
66
5,533
29 Jul 2025
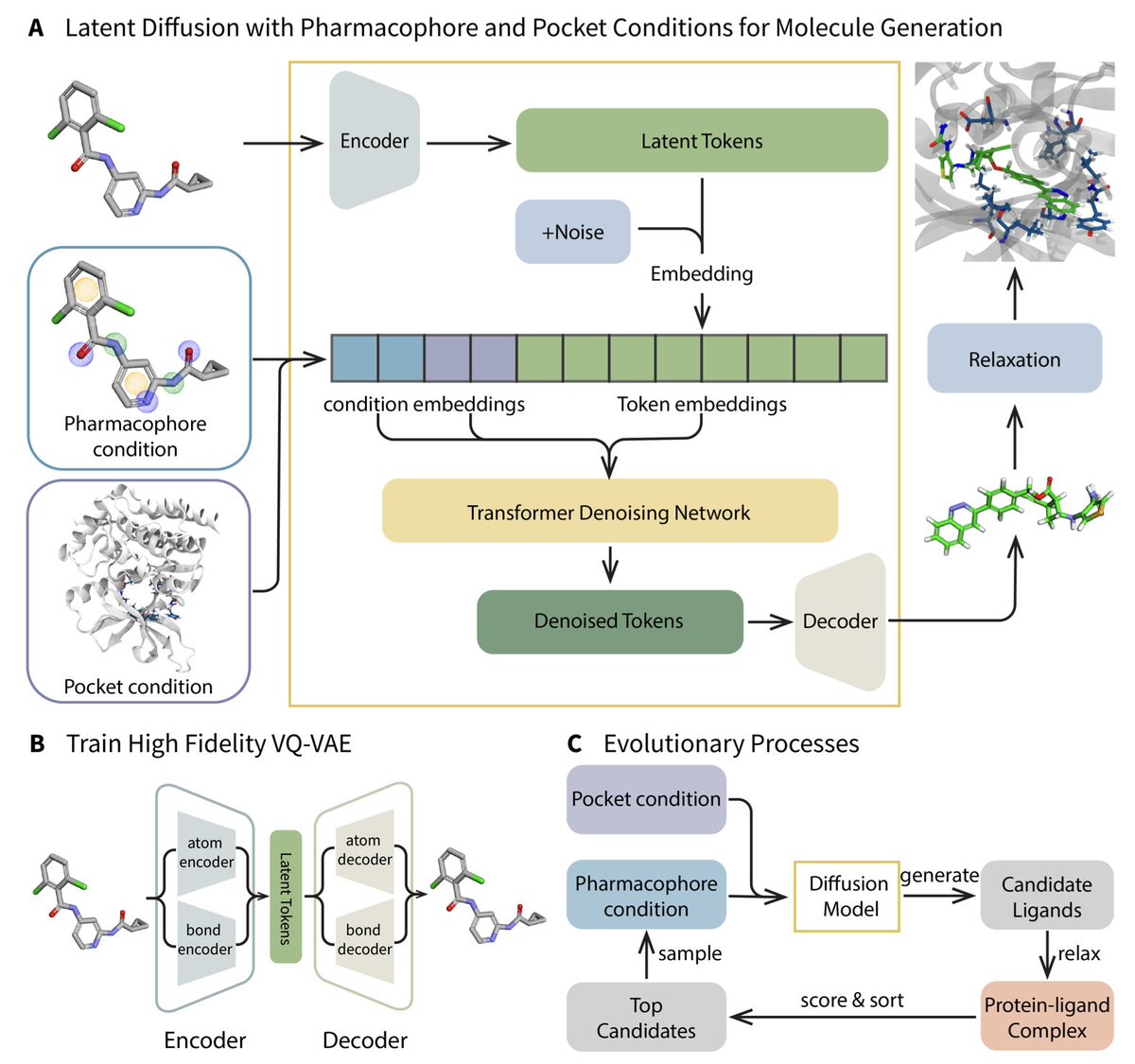
Generative Molecule Evolution Using 3D Pharmacophore for Efficient Structure-Based Drug Design
1. A groundbreaking framework named MEVO has been introduced to bridge the gap between large-scale small molecule datasets and limited protein-ligand complex datasets, significantly enhancing the training data for structure-based drug design (SBDD) models. This innovative approach leverages pharmacophore-guided molecule generation and evolutionary optimization strategies to design high-affinity binders for various protein targets.
2. MEVO incorporates three key components: a high-fidelity VQ-VAE for molecule representation in latent space, a diffusion model for pharmacophore-guided molecule generation, and a pocket-aware evolutionary strategy for optimizing molecules with physics-based scoring functions. This combination allows for the efficient generation of molecules that fit both pharmacophore principles and target-binding constraints.
3. The framework demonstrates remarkable versatility and generalizability, validated through the design of potent inhibitors for KRASG12D, a challenging cancer target. MEVO-generated molecules exhibit binding affinities comparable to known highly active inhibitors, as evaluated by free energy perturbation (FEP) calculations.
4. MEVO effectively addresses the limitations of current SBDD methods by integrating extensive training data from ligand-only datasets, which are almost unlimited compared to protein-ligand complex datasets. This strategy allows the model to learn robust pharmacophore patterns and generate diverse and plausible molecules.
5. The evolutionary optimization strategy in MEVO uses a physics-informed scoring function to iteratively improve the binding affinity of generated molecules. This training-free approach can be easily transferred to other tasks, such as optimizing ligand properties like permeability and solubility.
6. MEVO generates drug-like molecules with desirable properties, as evidenced by comparisons with FDA-approved drugs. The generated molecules comply with Lipinski’s rule of five, exhibit reasonable drug-likeness scores, and have similar logP distributions to reference molecules.
7. In hit discovery, MEVO successfully identifies high-affinity binders for various protein targets, outperforming known binders in some cases. The model’s ability to preserve critical interactions within binding pockets provides a strong foundation for developing potent ligands.
8. MEVO’s high versatility is showcased through its application in different stages of drug design, including ligand growing, linker design, and lead optimization. The model’s ability to design high-affinity binders for KRASG12D highlights its potential in tackling challenging drug targets.
📜Paper: arxiv.org/abs/2507.20130
#DrugDesign #GenerativeModels #StructureBasedDrugDesign #Pharmacophore #EvolutionaryOptimization #KRASInhibitors #AIinPharma
7
1,236

21 Apr 2023
#notablepaper
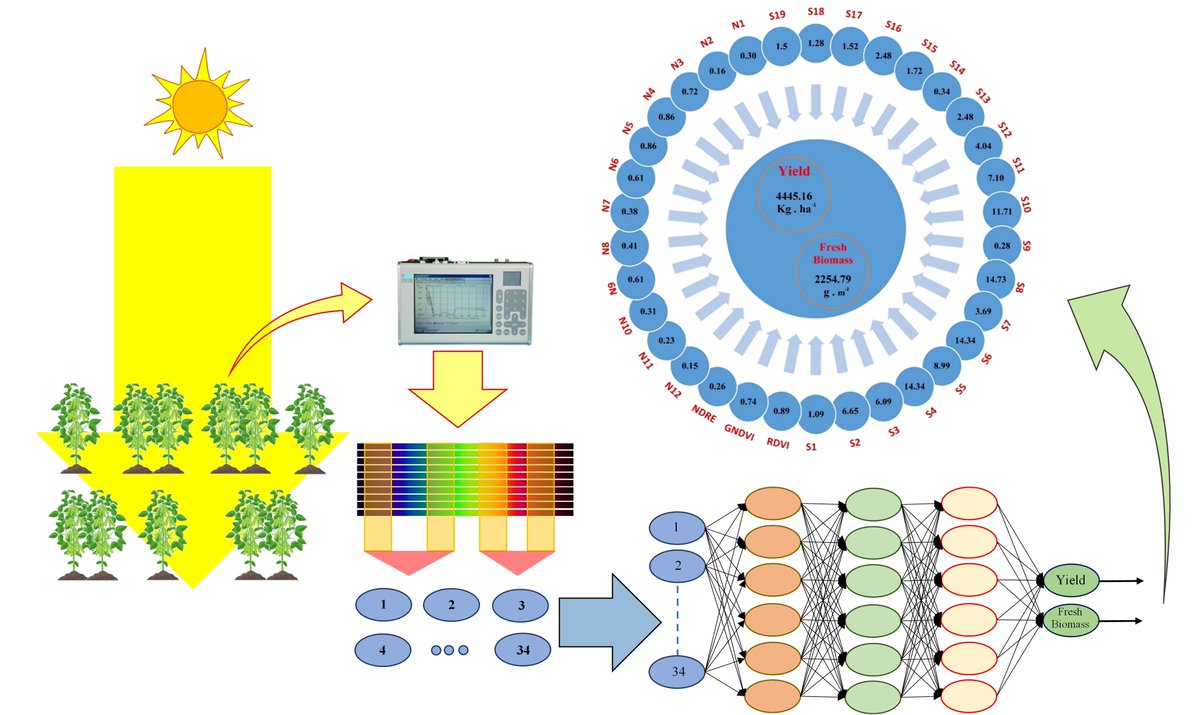
📢 Using Hybrid #ArtificialIntelligence and #EvolutionaryOptimization Algorithms for Estimating #SoybeanYield and #FreshBiomass Using #Hyperspectral #VegetationIndices
by Mohsen Yoosefzadeh-Najafabadi, Dan Tulpan, and Milad Eskandari
🔗mdpi.com/2072-4292/13/13/255…
1
5
1,268

14 Feb 2022
#Openaccess #paper by Luis M. Montero, Antonio Bello & Javier Reneses, researchers at @IIT_Comillas @Comillas_ICAI @UCOMILLAS, et al.
mdpi.com/1996-1073/15/4/1296
#unitcommitment #optimalthermalgeneration #numericaloptimization #evolutionaryoptimization #optimizationtechniques
1
4

11 Aug 2021
#Genetic programming guidance control system for a #reentry vehicle under #uncertainties
strathprints.strath.ac.uk/77… #OpenAccess #DifferentialEvolution #EvolutionaryOptimization @MAE_Strath @fmarchetti17 @edmondo_minisci
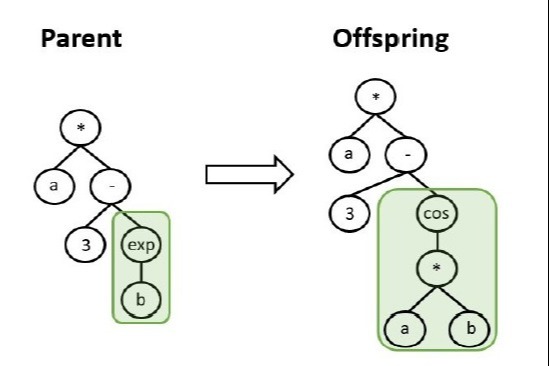
3
2

28 Apr 2020
[Article] Research and Study of the Hybrid Algorithms Based on the Collective Behavior of Fish Schools and Classical Optimization Methods
mdpi.com/1999-4893/13/4/85
#evolutionaryoptimization
#swarmintelligence
#fishschoolsearch
#gradientdescent
#hybridalgorithm
#algorithms

1
1

10 Aug 2018
Our CTO, Risto Miikkulainen, discusses how we discover novel solutions using #EvolutionaryOptimization on the Future Tech Podcast. Listen here: ow.ly/PNIk30lmf7V @FuturetechP

1
5

10 Aug 2018
@SentientDAI Discovering #NovelSolutions Via #EvolutionaryOptimization. Listen to it here: goo.gl/9JRiUU

1
2

11 May 2016
Open for Submissions #EvolutionaryOptimization #RoboticsIntelligence
#SelfOrganisingSystems fron.tiers.in/go/FNg2Vr

3
2

1 Mar 2016
A very nice introduction to particle swarms. #ArtificialIntelligence #evolutionaryoptimization #ai #geneticalgorithm


18 Dec 2015
Fantastic GA is efficient for feature selection #evolutionaryoptimization #machinelearning #datascience
16 Dec 2015

Genetic Algorithms: power tool for #DataScience modeling & #MachineLearning
linkedin.com/pulse/revolutio… by @manishpedia

2
1
16 Dec 2015
Using #evolutionarycomputation #evolutionaryoptimization on Financial #sentimentdata #datascience #machinelearning
22 Apr 2015

Building rules using #evolutionarycomputation. Like #neuralnetworks benefits from increase in computational power.
2
4