Multi-scale Graph Autoregressive Modeling: Molecular Property Prediction via Next Token Prediction
1. This paper introduces Connection-Aware Motif Sequencing (CamS), a novel graph-to-sequence representation that enables decoder-only Transformers to learn molecular graphs through standard next-token prediction (NTP). CamS bridges the gap between SMILES-based NTP and graph-native masked modeling by serializing molecular graphs into structure-rich causal sequences, achieving state-of-the-art performance on MoleculeNet and MoleculeACE benchmarks.
2. CamS mines data-driven connection-aware motifs and serializes them via scaffold-rooted breadth-first search (BFS) to establish a stable core-to-periphery order. It concatenates sequences from fine to coarse motif scales, allowing the model to condition global scaffolds on dense, uncorrupted local structural evidence. This hierarchical modeling effectively drives attention toward subtle structural edits that determine activity cliffs.
3. The authors instantiate CamS-LLaMA by pre-training a vanilla LLaMA backbone on CamS sequences. Despite relying on minimal priors, CamS-LLaMA outperforms both SMILES-based language models and strong graph baselines. The interpretability analysis confirms that the multi-scale causal serialization improves attention focus on activity-determining structural edits, making the model more sensitive to fine-grained changes in molecular structure.
4. The study demonstrates that CamS resolves the conflict between graph topology preservation and scalable autoregressive training. It provides a unified graph-to-causal-sequence interface that makes molecular graphs directly learnable by standard decoder-only NTP. This approach not only enhances general-purpose representation strength but also boosts discriminability for activity-cliff prediction, where small local motif edits trigger large property shifts.
5. The experimental results show that CamS-LLaMA achieves overall state-of-the-art performance on MoleculeNet and MoleculeACE. On MoleculeNet, it improves average AUROC in classification and average RMSE in regression compared to previous best baselines. On MoleculeACE, it achieves the best average RMSE, outperforming all ML baselines on most tasks. The ablation studies further confirm that multi-scale concatenation is the key driver of performance.
📜Paper: arxiv.org/abs/2601.02530v1
#MolecularPropertyPrediction #Transformer #GraphRepresentation #NextTokenPrediction #CamS #MoleculeNet #MoleculeACE

2
19
1,952

1 Jul 2025
Embedded Morgan Fingerprints for more efficient molecular property predictions with machine learning
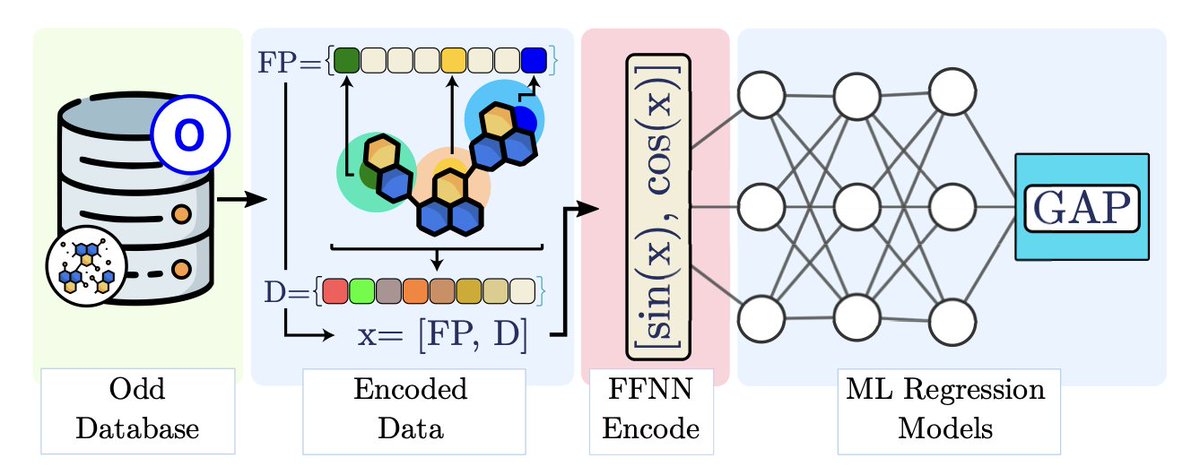
1.This work introduces Embedded Morgan Fingerprints (eMFP), a compact representation of traditional Morgan Fingerprints (MFP) designed to improve both speed and accuracy in molecular property prediction using machine learning.
2.eMFP reduces the dimensionality of high-bit MFP vectors by applying an embedding technique inspired by one-hot encoding compression. This helps mitigate overfitting and accelerates training without compromising molecular structural information.
3.Compared to MFP, eMFP achieves faster training and better prediction accuracy across various models including Random Forest, MLP, K-Neighbors Regressor, Gradient Boosted Trees, and Deep Neural Networks.
4.Evaluated across three datasets—RedDB, NFA, and QM9—the eMFP outperformed standard MFP in regression tasks predicting HOMO-LUMO gaps, especially for large datasets like QM9.
5.The optimal compression factors for eMFP were q = 16 and q = 32 for small/medium datasets, and q = 16 and q = 64 for large datasets, striking a balance between compactness and model performance.
6.eMFP retains the essential features of MFP, as confirmed through Principal Component Analysis and KDE of predictions. The structural integrity of the encoded data is preserved even at high compression.
7.Regression models trained with eMFP achieved higher R² values and narrower residual distributions (FWHM), indicating improved generalization and more consistent prediction quality.
8.Training time was significantly reduced using eMFP—up to several orders of magnitude faster than MFP—making it a practical choice under computational constraints and in large-scale modeling tasks.
9.eMFP enabled more extensive hyperparameter optimization within fixed time limits, leading to better model tuning. In contrast, MFP often failed to complete full optimization runs on large datasets like QM9.
10.This work highlights the potential of embedding techniques not just for cheminformatics, but also as a general strategy for compressing high-dimensional categorical data in ML workflows.
11.Overall, eMFP offers a more efficient, scalable, and often superior alternative to MFP, especially valuable for tasks requiring large datasets or fast model iteration.
💻Code: github.com/MMLabCodes/eMFP
📜Paper: doi.org/10.26434/chemrxiv-20…
#MachineLearning #Cheminformatics #MolecularML #Fingerprinting #DimensionalityReduction #DeepLearning #QM9 #OpenSource #GraphRepresentation #ComputationalChemistry
4
604

26 Nov 2024
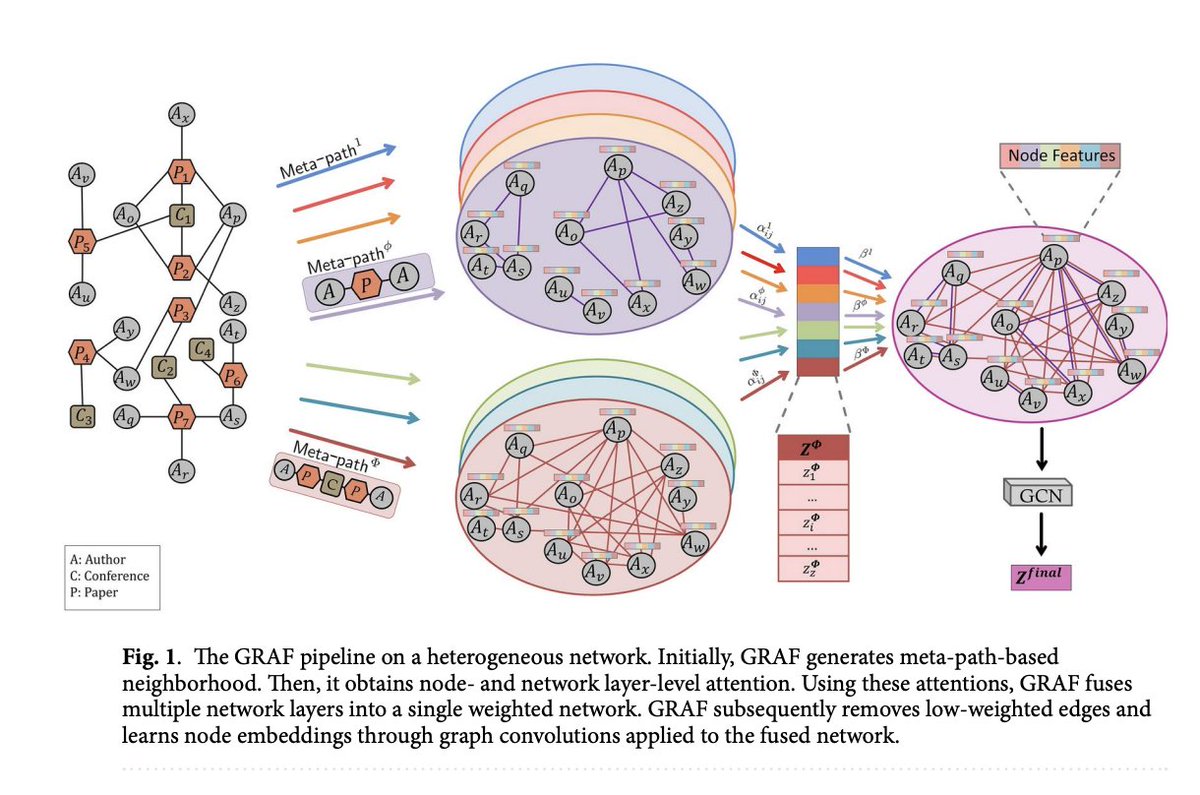
GRAF: A Machine Learning Framework that Convert Multiplex Heterogeneous Networks to Homogeneous Networks to Make Them more Suitable for Graph Representation Learning
itinai.com/graf-a-machine-le…
#GRAF #GraphRepresentation #NetworkAnalysis #MachineLearning #AIInnovation #ai #news #…
1
2
33
10 Nov 2024
Exploring Hierarchical Molecular Graph Representation in Multimodal LLMs
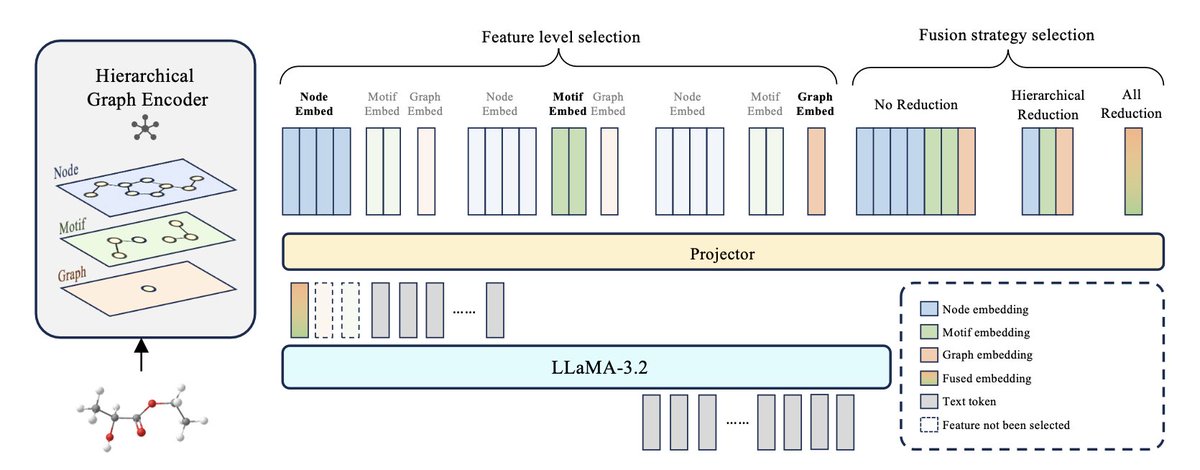
1. This study develops a hierarchical molecular graph encoder within a multimodal large language model (MLLM), enabling the integration of multi-level graph features, including node, motif, and graph-level information. This approach provides fine-grained molecular representations that enhance the MLLM’s capacity for complex biochemical tasks.
2. The model employs a multi-level segmentation pipeline that captures and aligns graph-based features with text, improving downstream performance on molecular tasks like reaction prediction, molecular property prediction, and molecular description generation.
3. Key insights from the study reveal that different molecular tasks benefit from distinct feature levels. For example, graph-level features are most effective for reaction prediction, while motif-level features excel in molecular description tasks, suggesting a need for dynamic processing to optimize task-specific performance.
4. The work also explores three feature fusion strategies—no reduction, hierarchical reduction, and all reduction—finding that task performance varies depending on the fusion method, with no-reduction consistently performing best in high-granularity tasks.
5. This hierarchical encoding framework lays the groundwork for future research in MLLMs for chemistry, highlighting the importance of multi-level graph representation to enhance molecular understanding and prediction accuracy.
📜Paper: arxiv.org/abs/2411.04708
#MolecularML #GraphRepresentation #MultimodalLLM #Bioinformatics #ChemicalReactions
1
3
12
1,994
5 Nov 2024
UniMAP: Universal SMILES-Graph Representation Learning
1. The UniMAP model introduces a universal approach for molecular representation learning, combining SMILES strings and molecular graphs to capture fine-grained “semantics” and improve property predictions.
2. UniMAP innovates by integrating SMILES and graph data, resolving issues in previous single-modality models that struggled with subtle sequence/graph differences affecting molecular properties.
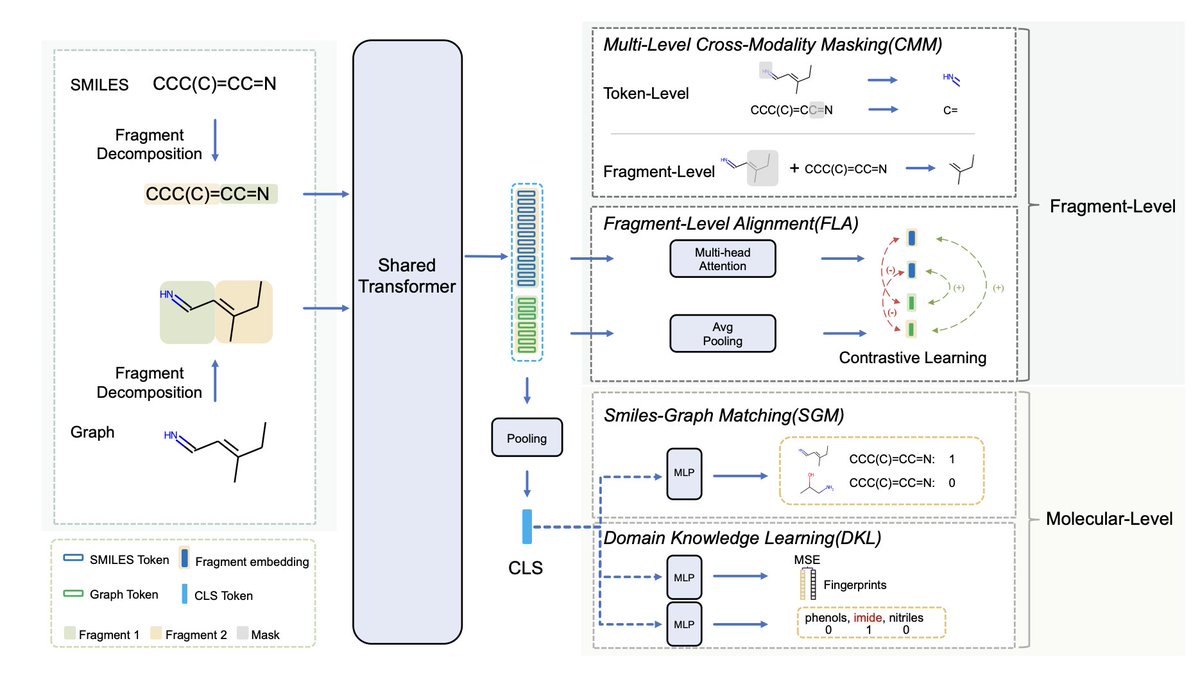
3. Utilizing a multi-layer Transformer, UniMAP conducts deep cross-modality fusion with four key pre-training tasks: Multi-Level Cross-Modality Masking (CMM), SMILES-Graph Matching (SGM), Fragment-Level Alignment (FLA), and Domain Knowledge Learning (DKL).
4. With CMM and FLA tasks, UniMAP captures both global and local alignment between modalities, allowing for a comprehensive representation and enhancing the model’s accuracy in property predictions.
5. Pre-trained on 10 million molecules, UniMAP demonstrates superior performance in downstream tasks like molecular property prediction, drug-target affinity, and drug-drug interaction, outperforming current pre-trained models in various benchmarks.
6. Visualization of UniMAP embeddings reveals clear clustering based on molecular scaffolds and fragment types, showing UniMAP’s ability to recognize chemically relevant structures across modalities.
7. The UniMAP approach has significant implications for drug discovery, offering an efficient and accurate model that combines SMILES and graph data to enhance prediction capabilities in cheminformatics and bioinformatics.
💻Code: anonymous.4open.science/r/Un…
📜Paper: arxiv.org/abs/2310.14216v2
#MolecularRepresentation #MachineLearning #DrugDiscovery #SMILES #GraphRepresentation #Transformer
8
1,393

13 Jun 2021
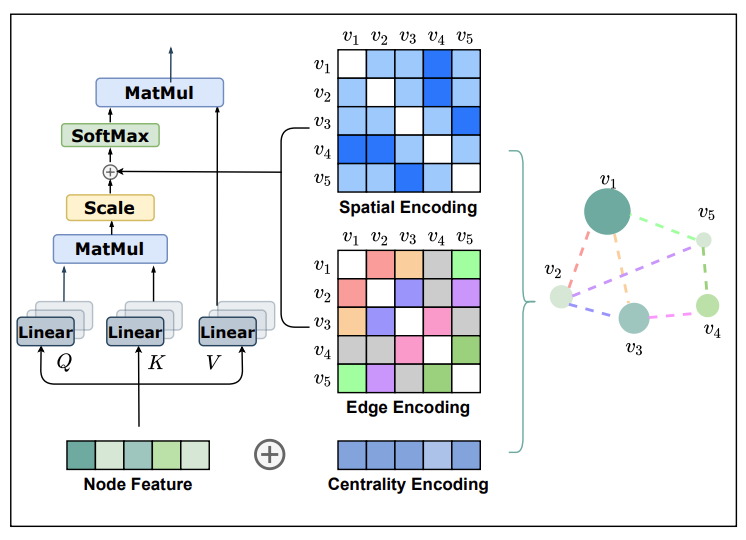
Do Transformers Really Perform Bad for Graph Representation?
Ying et al.: arxiv.org/abs/2106.05234
#Transformers #GraphRepresentation #DeepLearning
4
5

6 Oct 2020
Great to see great works in #MICCAI2020 oral sessions about Functional Brain Networks and Neuroimaging!!!
#DeepLearning #connectome #graphrepresentation #graphtheory #brainnetwork @MICCAI2020 @MiccaiStudents @WomenInMICCAI

2
1
14

Check out a fresh approach to understanding the shape of large-scale data, which quickly and efficiently characterizes a dataset using the spectrum of its #GraphRepresentation. Learn more and grab the code below: goo.gle/3c8CqLs
1
60
224

20 Dec 2019
(2) Another paper is a new approach based on autoregressive flows for molecular graph generation and optimization openreview.net/forum?id=S1es… #graphrepresentation #drugdiscovery
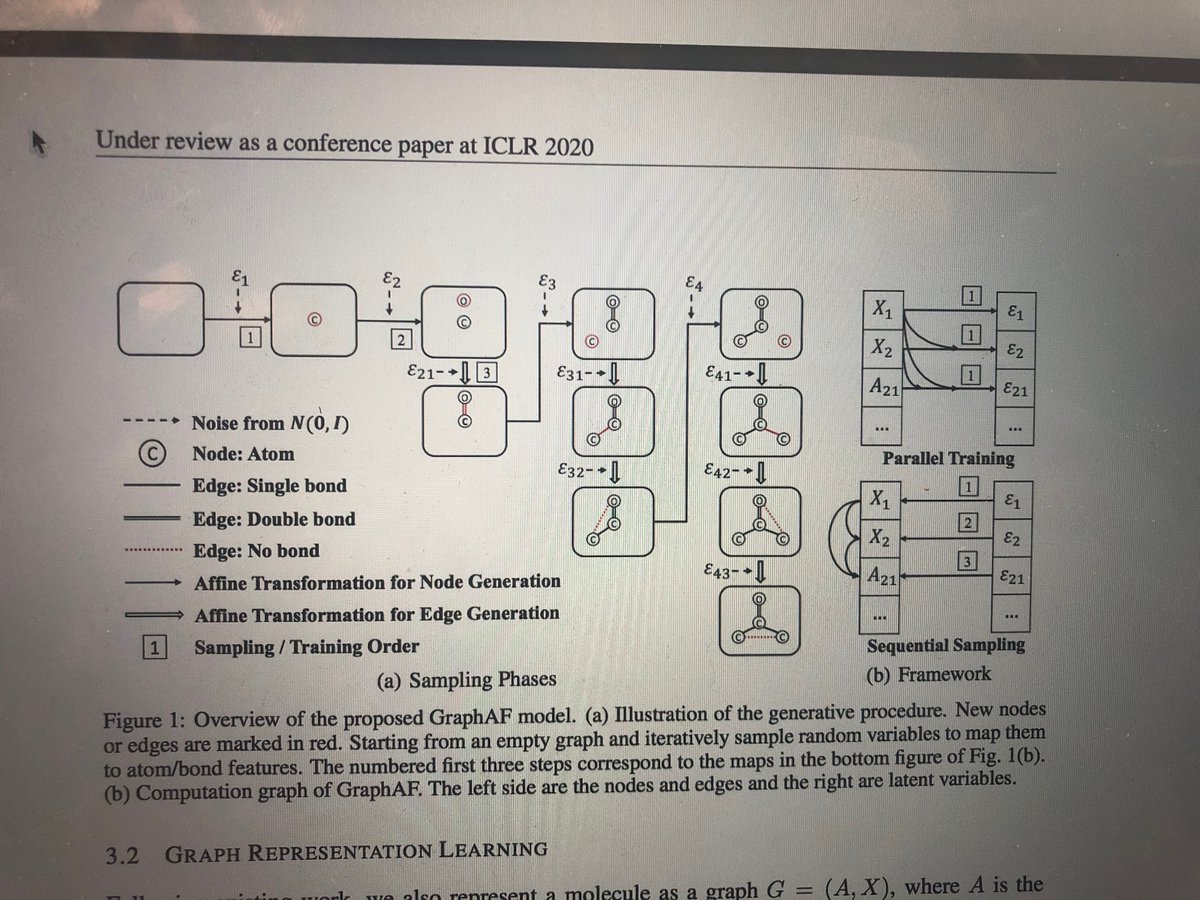
1
2
8
20 Dec 2019
Excited to share that two papers on drug discovery have been accepted to ICLR'20. (1) one is a new unsupervised and semi-supervised approach for learning graph-level representations for #molecule properties prediction. openreview.net/forum?id=r1lf… #graphrepresentation #drugdiscovery
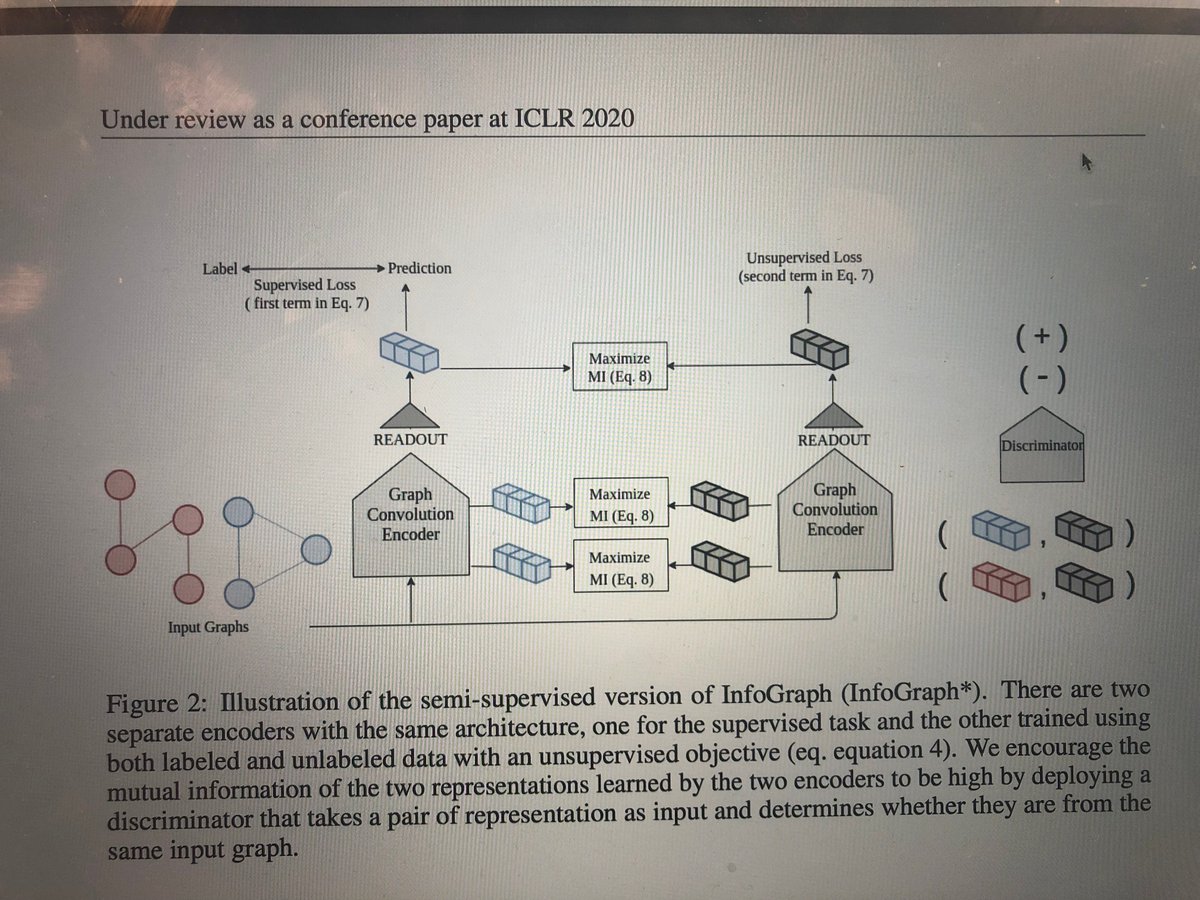
1
5
35