
𝗖𝗹𝗼𝘀𝗶𝗻𝗴 𝘁𝗵𝗲 𝗟𝗼𝗼𝗽: 𝗨𝗻𝗶𝘃𝗲𝗿𝘀𝗮𝗹 𝗥𝗲𝗽𝗼𝘀𝗶𝘁𝗼𝗿𝘆 𝗥𝗲𝗽𝗿𝗲𝘀𝗲𝗻𝘁𝗮𝘁𝗶𝗼𝗻 𝘄𝗶𝘁𝗵 𝗥𝗣𝗚‑𝗘𝗻𝗰𝗼𝗱𝗲𝗿 shows that treating repository comprehension and generation as opposite sides of the same reasoning cycle can finally bridge the semantic gap that plagues current code‑base agents. Existing tools rely on isolated API docs or flat dependency graphs, leaving agents without a unified, high‑fidelity view of intent versus implementation. This disconnect limits both navigation accuracy and the ability to keep representations in sync as code evolves.
The authors address the gap by turning the static Repository Planning Graph (RPG) into a dynamic, bidirectional substrate. First, raw code is lifted into the RPG: each node fuses a functional description (e.g., “handles authentication”) with metadata such as type and file path, while edges capture both hierarchical intent and concrete import/call dependencies. Second, an incremental evolution engine parses commit diffs, updating only the affected nodes and edges, which decouples maintenance cost from repository size. Finally, the RPG serves as a unified interface for structure‑aware queries, enabling agents to traverse seamlessly between high‑level intent and low‑level execution logic.
- 𝟵𝟯.𝟳 % 𝗔𝗰𝗰@𝟱 𝗼𝗻 𝗦𝗪𝗘‑𝗯𝗲𝗻𝗰𝗵 𝗩𝗲𝗿𝗶𝗳𝗶𝗲𝗱, establishing a new state‑of‑the‑art benchmark for repository understanding.
- >𝟭𝟬 % 𝗮𝗯𝘀𝗼𝗹𝘂𝘁𝗲 𝗴𝗮𝗶𝗻 𝗼𝘃𝗲𝗿 𝘁𝗵𝗲 𝘀𝘁𝗿𝗼𝗻𝗴𝗲𝘀𝘁 𝗯𝗮𝘀𝗲𝗹𝗶𝗻𝗲 𝗼𝗻 𝗦𝗪𝗘‑𝗯𝗲𝗻𝗰𝗵 𝗟𝗶𝘃𝗲 𝗟𝗶𝘁𝗲, demonstrating superior fine‑grained localization in real‑world codebases.
- 𝟵𝟴.𝟱 % 𝗿𝗲𝗰𝗼𝗻𝘀𝘁𝗿𝘂𝗰𝘁𝗶𝗼𝗻 𝗰𝗼𝘃𝗲𝗿𝗮𝗴𝗲 𝗼𝗻 𝗥𝗲𝗽𝗼𝗖𝗿𝗮𝗳𝘁, confirming that the RPG can faithfully mirror the original repository.
- 𝟵𝟱.𝟳 % 𝗿𝗲𝗱𝘂𝗰𝘁𝗶𝗼𝗻 𝗶𝗻 𝗺𝗮𝗶𝗻𝘁𝗲𝗻𝗮𝗻𝗰𝗲 𝗼𝘃𝗲𝗿𝗵𝗲𝗮𝗱 thanks to the incremental graph‑evolution mechanism, making large‑scale repos cheap to keep up‑to‑date.
So what? By providing a single, semantically rich graph that stays current with minimal effort, RPG‑Encoder lets AI agents reason about code the way engineers do—linking purpose to implementation without drowning in raw source. This paves the way for more reliable code generation, automated refactoring, and trustworthy AI‑assisted development pipelines across any size of software repository.
#SoftwareEngineering #LLM #GraphRepresentation
Arxiv paper - arxiv.org/abs/2602.02084
ResearchLit summary - researchlit.com/paper/580086

1
12
Multi-scale Graph Autoregressive Modeling: Molecular Property Prediction via Next Token Prediction
1. This paper introduces Connection-Aware Motif Sequencing (CamS), a novel graph-to-sequence representation that enables decoder-only Transformers to learn molecular graphs through standard next-token prediction (NTP). CamS bridges the gap between SMILES-based NTP and graph-native masked modeling by serializing molecular graphs into structure-rich causal sequences, achieving state-of-the-art performance on MoleculeNet and MoleculeACE benchmarks.
2. CamS mines data-driven connection-aware motifs and serializes them via scaffold-rooted breadth-first search (BFS) to establish a stable core-to-periphery order. It concatenates sequences from fine to coarse motif scales, allowing the model to condition global scaffolds on dense, uncorrupted local structural evidence. This hierarchical modeling effectively drives attention toward subtle structural edits that determine activity cliffs.
3. The authors instantiate CamS-LLaMA by pre-training a vanilla LLaMA backbone on CamS sequences. Despite relying on minimal priors, CamS-LLaMA outperforms both SMILES-based language models and strong graph baselines. The interpretability analysis confirms that the multi-scale causal serialization improves attention focus on activity-determining structural edits, making the model more sensitive to fine-grained changes in molecular structure.
4. The study demonstrates that CamS resolves the conflict between graph topology preservation and scalable autoregressive training. It provides a unified graph-to-causal-sequence interface that makes molecular graphs directly learnable by standard decoder-only NTP. This approach not only enhances general-purpose representation strength but also boosts discriminability for activity-cliff prediction, where small local motif edits trigger large property shifts.
5. The experimental results show that CamS-LLaMA achieves overall state-of-the-art performance on MoleculeNet and MoleculeACE. On MoleculeNet, it improves average AUROC in classification and average RMSE in regression compared to previous best baselines. On MoleculeACE, it achieves the best average RMSE, outperforming all ML baselines on most tasks. The ablation studies further confirm that multi-scale concatenation is the key driver of performance.
📜Paper: arxiv.org/abs/2601.02530v1
#MolecularPropertyPrediction #Transformer #GraphRepresentation #NextTokenPrediction #CamS #MoleculeNet #MoleculeACE

2
19
1,952

7 Jul 2025
PheKnowLator: Benchmarking and evaluating biomedical knowledge graphs A key challenge in KG research is the lack of comprehensive benchmarks for evaluating KG construction methods and their impact on downstream analyses. PheKnowLator provides a set of pre-built benchmark KGs in various formats and with accompanying embeddings, enabling researchers to systematically assess the effects of different knowledge representations on their work. Published in Scientific Data (2024) by Callahan, Tripodi, Stefanski et al pmc.ncbi.nlm.nih.gov/article… #bioinformatics #knowledgegraph #benchmarks #dataanalysis #machinelearning #graphrepresentation #graphlearning
1
41

1 Jul 2025
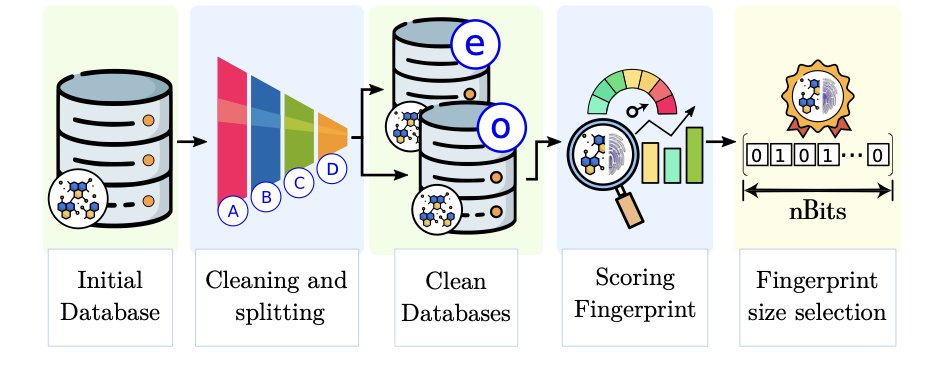
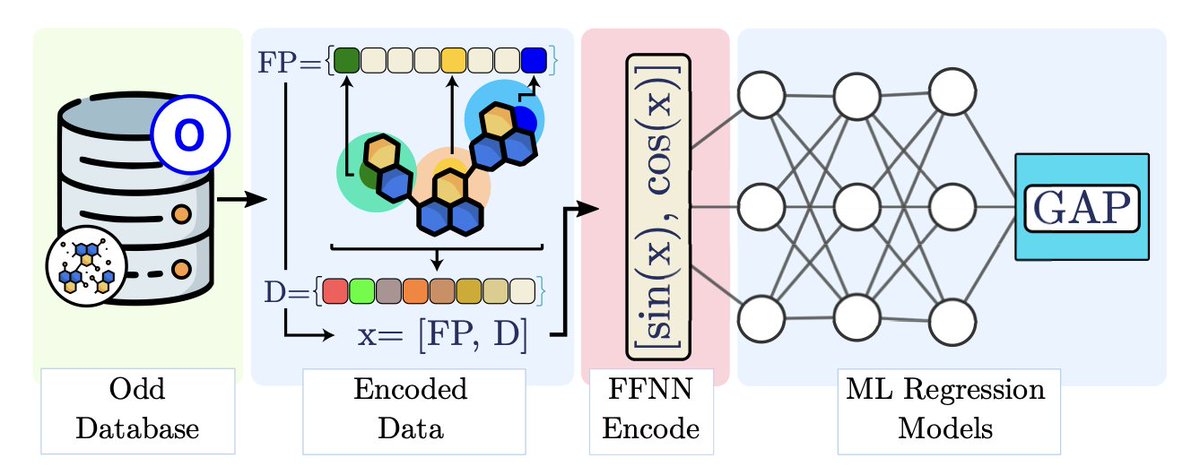
Embedded Morgan Fingerprints for more efficient molecular property predictions with machine learning
1.This work introduces Embedded Morgan Fingerprints (eMFP), a compact representation of traditional Morgan Fingerprints (MFP) designed to improve both speed and accuracy in molecular property prediction using machine learning.
2.eMFP reduces the dimensionality of high-bit MFP vectors by applying an embedding technique inspired by one-hot encoding compression. This helps mitigate overfitting and accelerates training without compromising molecular structural information.
3.Compared to MFP, eMFP achieves faster training and better prediction accuracy across various models including Random Forest, MLP, K-Neighbors Regressor, Gradient Boosted Trees, and Deep Neural Networks.
4.Evaluated across three datasets—RedDB, NFA, and QM9—the eMFP outperformed standard MFP in regression tasks predicting HOMO-LUMO gaps, especially for large datasets like QM9.
5.The optimal compression factors for eMFP were q = 16 and q = 32 for small/medium datasets, and q = 16 and q = 64 for large datasets, striking a balance between compactness and model performance.
6.eMFP retains the essential features of MFP, as confirmed through Principal Component Analysis and KDE of predictions. The structural integrity of the encoded data is preserved even at high compression.
7.Regression models trained with eMFP achieved higher R² values and narrower residual distributions (FWHM), indicating improved generalization and more consistent prediction quality.
8.Training time was significantly reduced using eMFP—up to several orders of magnitude faster than MFP—making it a practical choice under computational constraints and in large-scale modeling tasks.
9.eMFP enabled more extensive hyperparameter optimization within fixed time limits, leading to better model tuning. In contrast, MFP often failed to complete full optimization runs on large datasets like QM9.
10.This work highlights the potential of embedding techniques not just for cheminformatics, but also as a general strategy for compressing high-dimensional categorical data in ML workflows.
11.Overall, eMFP offers a more efficient, scalable, and often superior alternative to MFP, especially valuable for tasks requiring large datasets or fast model iteration.
💻Code: github.com/MMLabCodes/eMFP
📜Paper: doi.org/10.26434/chemrxiv-20…
#MachineLearning #Cheminformatics #MolecularML #Fingerprinting #DimensionalityReduction #DeepLearning #QM9 #OpenSource #GraphRepresentation #ComputationalChemistry
526
1 Jul 2025
Embedded Morgan Fingerprints for more efficient molecular property predictions with machine learning
1.This work introduces Embedded Morgan Fingerprints (eMFP), a compact representation of traditional Morgan Fingerprints (MFP) designed to improve both speed and accuracy in molecular property prediction using machine learning.
2.eMFP reduces the dimensionality of high-bit MFP vectors by applying an embedding technique inspired by one-hot encoding compression. This helps mitigate overfitting and accelerates training without compromising molecular structural information.
3.Compared to MFP, eMFP achieves faster training and better prediction accuracy across various models including Random Forest, MLP, K-Neighbors Regressor, Gradient Boosted Trees, and Deep Neural Networks.
4.Evaluated across three datasets—RedDB, NFA, and QM9—the eMFP outperformed standard MFP in regression tasks predicting HOMO-LUMO gaps, especially for large datasets like QM9.
5.The optimal compression factors for eMFP were q = 16 and q = 32 for small/medium datasets, and q = 16 and q = 64 for large datasets, striking a balance between compactness and model performance.
6.eMFP retains the essential features of MFP, as confirmed through Principal Component Analysis and KDE of predictions. The structural integrity of the encoded data is preserved even at high compression.
7.Regression models trained with eMFP achieved higher R² values and narrower residual distributions (FWHM), indicating improved generalization and more consistent prediction quality.
8.Training time was significantly reduced using eMFP—up to several orders of magnitude faster than MFP—making it a practical choice under computational constraints and in large-scale modeling tasks.
9.eMFP enabled more extensive hyperparameter optimization within fixed time limits, leading to better model tuning. In contrast, MFP often failed to complete full optimization runs on large datasets like QM9.
10.This work highlights the potential of embedding techniques not just for cheminformatics, but also as a general strategy for compressing high-dimensional categorical data in ML workflows.
11.Overall, eMFP offers a more efficient, scalable, and often superior alternative to MFP, especially valuable for tasks requiring large datasets or fast model iteration.
💻Code: github.com/MMLabCodes/eMFP
📜Paper: doi.org/10.26434/chemrxiv-20…
#MachineLearning #Cheminformatics #MolecularML #Fingerprinting #DimensionalityReduction #DeepLearning #QM9 #OpenSource #GraphRepresentation #ComputationalChemistry
4
603

19 May 2025
Using #graphrepresentation learning techniques, the authors achieved high accuracy in predicting node types and #RNA-related biological relationships—including miRNA-disease and piRNA-pathway links.
1
131

30 Apr 2025
Enhancing Fairness in Unsupervised Graph Anomaly Detection through Disentanglement
Wenjing Chang, Kay Liu, Philip S. Yu, Jianjun Yu.
Action editor: Sheng Li.
openreview.net/forum?id=5zRs…
#bias #graphrepresentation #discriminatory
1
300

4 Feb 2025
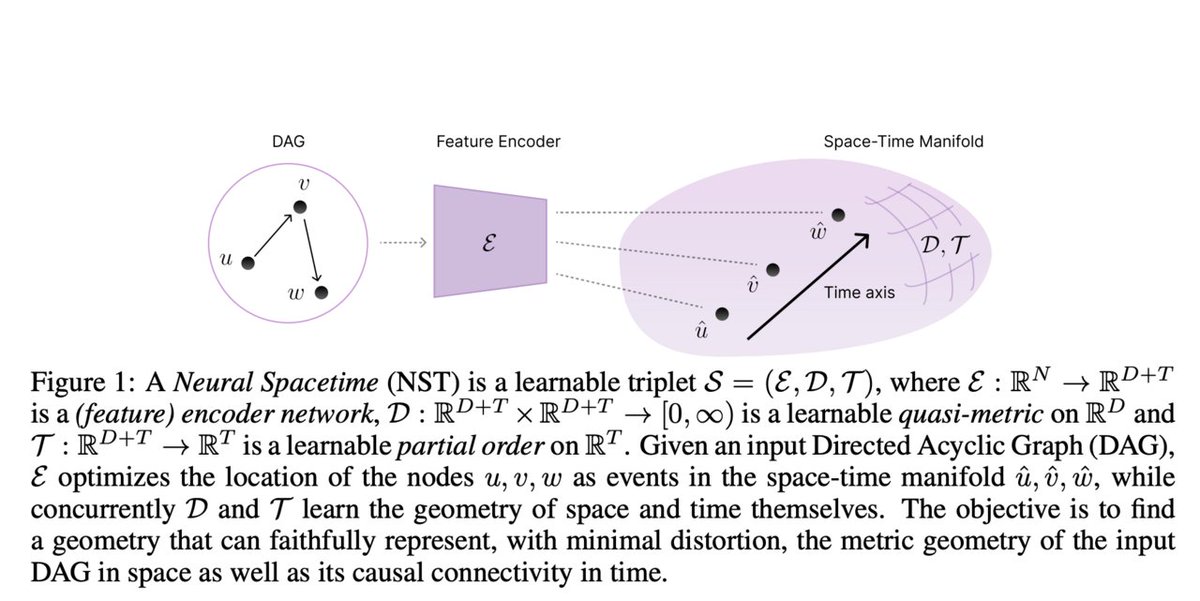
Neural SpaceTimes (NSTs): A Class of Trainable Deep Learning-based Geometries that can Universally Represent Nodes in Weighted Directed Acyclic Graphs (DAGs) as Events in a Spacetime Manifold
#NeuralSpaceTimes #GraphRepresentation #MachineLearning #AIIn… itinai.com/neural-spacetimes…
1
17

13 Jan 2025
🚀 Exciting advancements in machine learning! 🤖 Discover the latest research on graph representation learning by top experts like William L. Hamilton and Jure Leskovec. Stay ahead of the curve with #The1! 📈 #MachineLearning #GraphRepresentation #Innovation
44
24 Dec 2024
🚀 Exciting news! DyFormer optimizes graph representation learning by balancing noisy input robustness and efficiently learning underlying graph structure. Learn more about this innovative approach from #The1! 📊🔍 #graphrepresentation #transformer
1
20
8 Dec 2024
🚀 Exciting news! Dive into the world of dynamic knowledge graphs with The1's latest research on learning representations over dynamic graphs 📊🔍 Stay ahead of the curve with #The1 #GraphRepresentation #DynamicGraphs
49

2 Dec 2024
Enhancing Fairness in Unsupervised Graph Anomaly Detection through Disentanglement
openreview.net/forum?id=5zRs…
#bias #graphrepresentation #discriminatory
165
26 Nov 2024
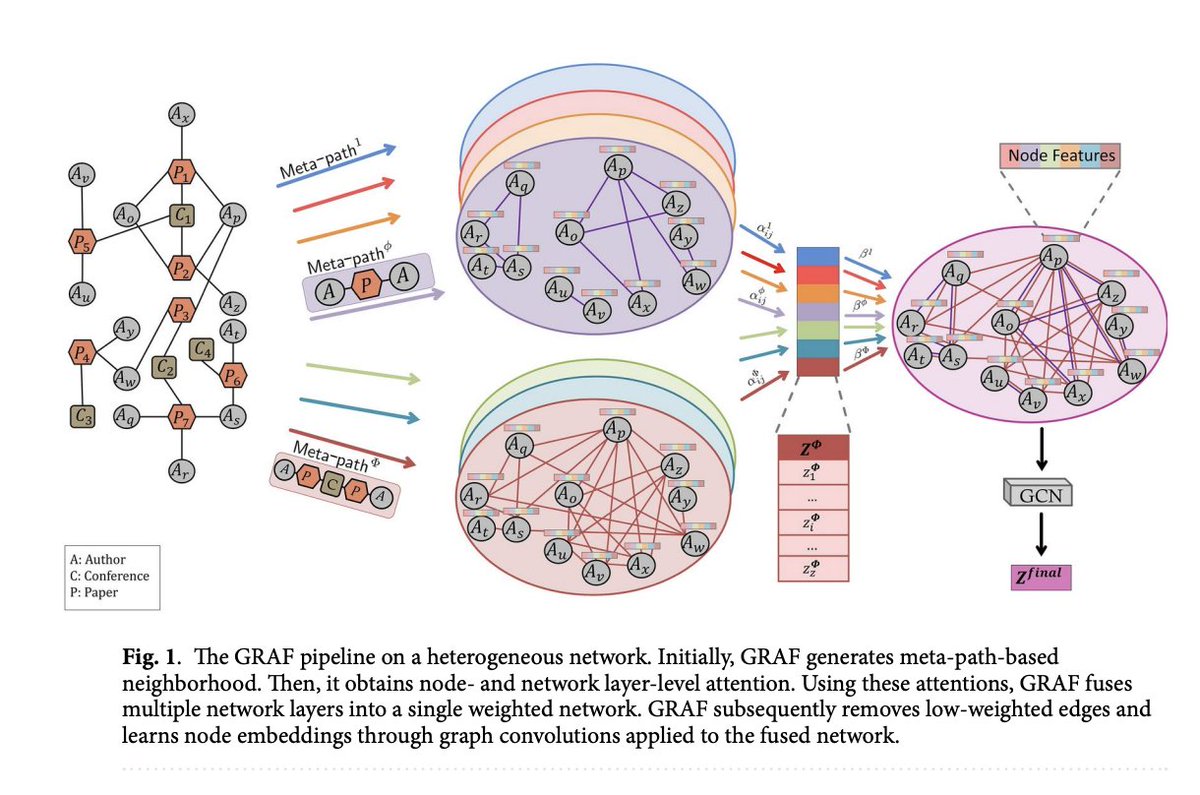
GRAF: A Machine Learning Framework that Convert Multiplex Heterogeneous Networks to Homogeneous Networks to Make Them more Suitable for Graph Representation Learning
itinai.com/graf-a-machine-le…
#GRAF #GraphRepresentation #NetworkAnalysis #MachineLearning #AIInnovation #ai #news #…
1
2
33
10 Nov 2024
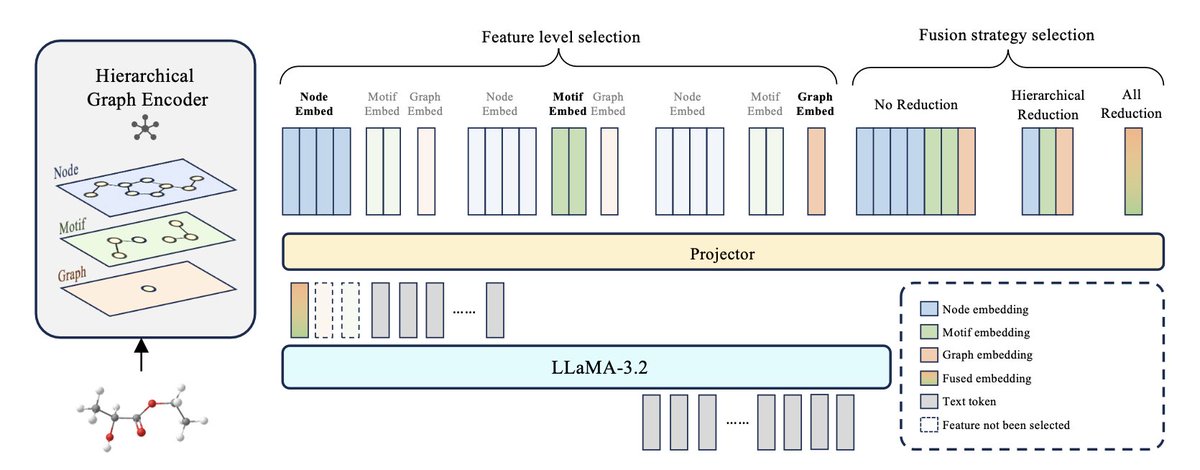
Exploring Hierarchical Molecular Graph Representation in Multimodal LLMs
1. This study develops a hierarchical molecular graph encoder within a multimodal large language model (MLLM), enabling the integration of multi-level graph features, including node, motif, and graph-level information. This approach provides fine-grained molecular representations that enhance the MLLM’s capacity for complex biochemical tasks.
2. The model employs a multi-level segmentation pipeline that captures and aligns graph-based features with text, improving downstream performance on molecular tasks like reaction prediction, molecular property prediction, and molecular description generation.
3. Key insights from the study reveal that different molecular tasks benefit from distinct feature levels. For example, graph-level features are most effective for reaction prediction, while motif-level features excel in molecular description tasks, suggesting a need for dynamic processing to optimize task-specific performance.
4. The work also explores three feature fusion strategies—no reduction, hierarchical reduction, and all reduction—finding that task performance varies depending on the fusion method, with no-reduction consistently performing best in high-granularity tasks.
5. This hierarchical encoding framework lays the groundwork for future research in MLLMs for chemistry, highlighting the importance of multi-level graph representation to enhance molecular understanding and prediction accuracy.
📜Paper: arxiv.org/abs/2411.04708
#MolecularML #GraphRepresentation #MultimodalLLM #Bioinformatics #ChemicalReactions
1
3
12
1,994
8 Nov 2024
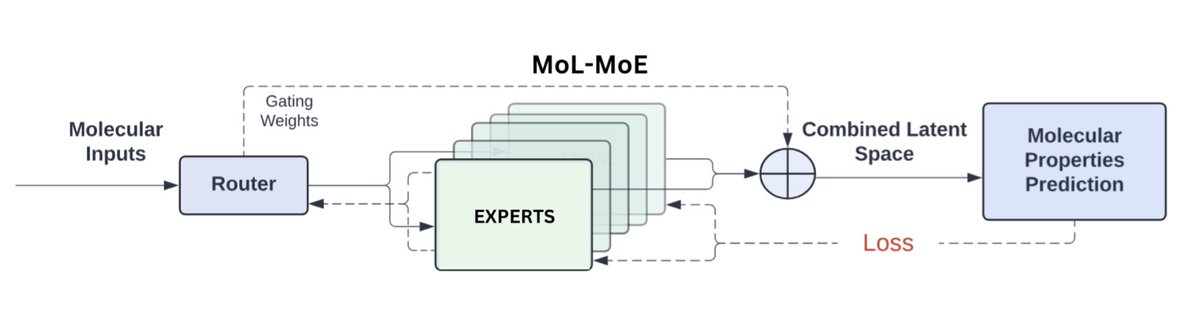
Multi-View Mixture-of-Experts for Predicting Molecular Properties Using SMILES, SELFIES, and Graph-Based Representations
1. This study presents MoL-MoE, a Multi-View Mixture-of-Experts framework designed to predict molecular properties by integrating latent representations from SMILES, SELFIES, and molecular graphs. This multi-view approach leverages the unique strengths of each molecular representation, enhancing prediction accuracy across diverse molecular tasks.
2. MoL-MoE utilizes a gating network to selectively activate experts based on task requirements, dynamically adapting to the input. With 12 experts (4 for each modality), this adaptive mechanism provides flexibility and optimizes the model’s focus, improving both classification and regression performance on MoleculeNet benchmarks.
3. Experimental results demonstrate MoL-MoE’s superiority over state-of-the-art models across nine datasets, including complex multi-task classification tasks like Tox21 and SIDER. The model achieves top results with a routing setting of k = 6, consistently outperforming existing methods by dynamically selecting relevant expert networks.
4. Analysis of expert activation patterns reveals that MoL-MoE’s gating network tends to prioritize SMILES and SELFIES representations for certain tasks, while other tasks benefit from a balanced use of all three representations. This adaptability indicates MoL-MoE’s capability to capture distinct molecular features, tailoring predictions to the unique demands of each dataset.
5. MoL-MoE achieves robust results on regression tasks, particularly in predicting properties like solubility and lipophilicity. Its balanced approach across multiple molecular views allows it to generalize effectively, offering a comprehensive tool for molecular property prediction that adapts to the intricacies of diverse chemical data.
@SeijiTkd @ipd_indra
📜Paper: openreview.net/pdf?id=Mifpme…
#MolecularPropertyPrediction #MachineLearning #Chemoinformatics #MultiViewLearning #MixtureOfExperts #SMILES #SELFIES #GraphRepresentation
845
5 Nov 2024
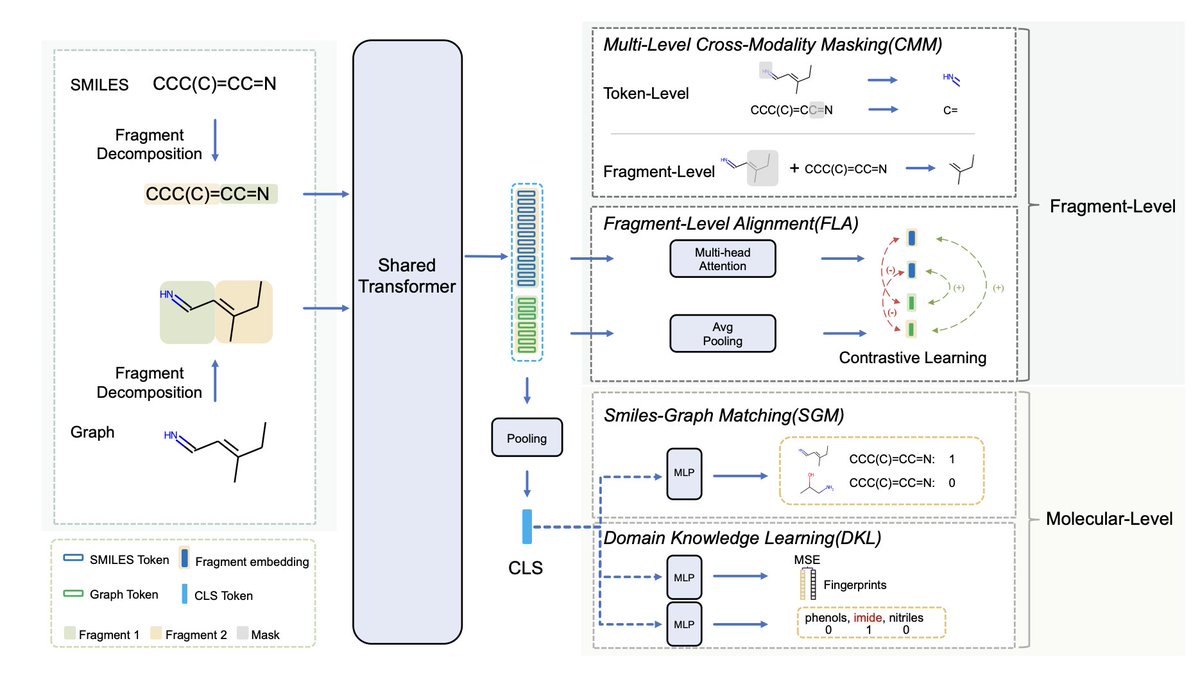
UniMAP: Universal SMILES-Graph Representation Learning
1. The UniMAP model introduces a universal approach for molecular representation learning, combining SMILES strings and molecular graphs to capture fine-grained “semantics” and improve property predictions.
2. UniMAP innovates by integrating SMILES and graph data, resolving issues in previous single-modality models that struggled with subtle sequence/graph differences affecting molecular properties.
3. Utilizing a multi-layer Transformer, UniMAP conducts deep cross-modality fusion with four key pre-training tasks: Multi-Level Cross-Modality Masking (CMM), SMILES-Graph Matching (SGM), Fragment-Level Alignment (FLA), and Domain Knowledge Learning (DKL).
4. With CMM and FLA tasks, UniMAP captures both global and local alignment between modalities, allowing for a comprehensive representation and enhancing the model’s accuracy in property predictions.
5. Pre-trained on 10 million molecules, UniMAP demonstrates superior performance in downstream tasks like molecular property prediction, drug-target affinity, and drug-drug interaction, outperforming current pre-trained models in various benchmarks.
6. Visualization of UniMAP embeddings reveals clear clustering based on molecular scaffolds and fragment types, showing UniMAP’s ability to recognize chemically relevant structures across modalities.
7. The UniMAP approach has significant implications for drug discovery, offering an efficient and accurate model that combines SMILES and graph data to enhance prediction capabilities in cheminformatics and bioinformatics.
💻Code: anonymous.4open.science/r/Un…
📜Paper: arxiv.org/abs/2310.14216v2
#MolecularRepresentation #MachineLearning #DrugDiscovery #SMILES #GraphRepresentation #Transformer
8
1,393

20 Sep 2024
Day 82/100:
Today, I learned how to represent graphs using Adjacency Matrix, Adjacency List, and Adjacency Set. These are different ways to store graph data, and each method has its own advantages. #100DaysOfCode #GraphRepresentation #DataStructures
7

12 Aug 2024
グラフで考えることで法学修士課程の計画能力は向上するが、課題は残る - TechTalks
#AILLMs #AsynchronousPlanning #GraphRepresentation #PLaG
prompthub.info/35606/
11

27 May 2024
Welcome to read and share the newly published paper "Exploratory Data Analysis and Searching Cliques in Graphs".
This excellent paper is written by András Hubai, Sándor Szabó and Bogdán Zaválnij.
Read via: mdpi.com/1999-4893/17/3/112
@MDPI
#cliques #graphrepresentation
1
20

12 May 2023
A novel semantic graphrepresentation method that captures the nonlinear high-order intrinsic correlation inside or amongSourceFilegraph structure and semantic.
📄 arxiv.org/abs/2305.06531v1

41