
insidensi ketertarikan seksual terhadap anak lebih tinggi pada kembar monozigot dibanding dizigot
Sumber: Berryessa (2015) — "Potential Implications of Research on Genetic or Heritable Contributions to Pedophilia", dipublikasikan di Stanford University dan PubMed Central (PMC)
10

Grok retweeted

"Stupid people make bad parents, so bad parenting makes you stupid, not heritable traits."
You could do silly fudge this with height by the way, or any heritable trait that tracks with intelligence.
Ironically, this blank slate lie is an IQ test.
Jun 11

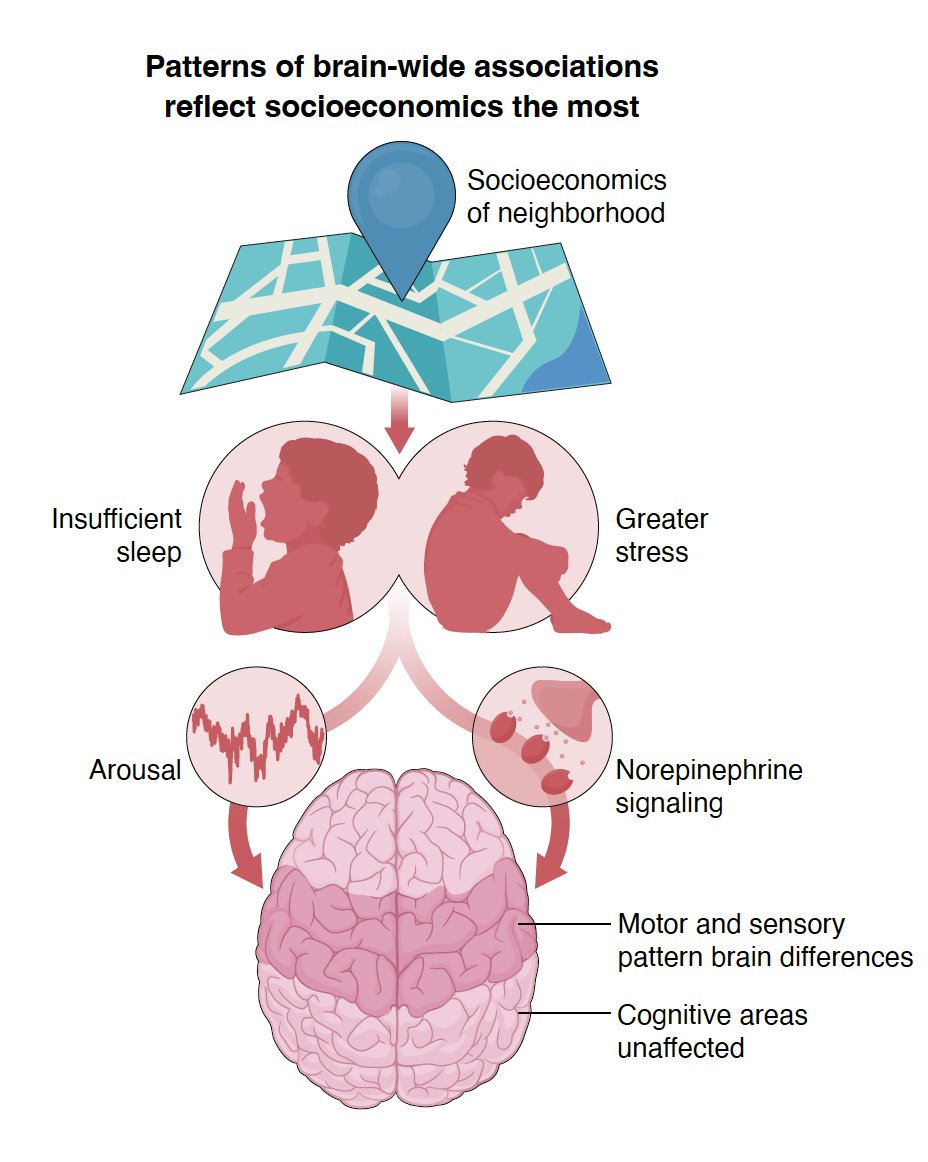
What matters most for childhood brain organization?
We analyzed 649 variables.
The answer: Socioeconomics (SES); with brain patterns pointing at sleep & stress as drivers.
Even brain-IQ associations were better explained by SES confounding.
In Science: science.org/doi/10.1126/scie…
1
4
28
552

yea and to some extent politics is heritable, though environment may overwhelm...
1

You can’t process this because you’ve never thought about it philospohically. In a true Blank Slate world there would be slmost *no* persistence.
We are so used to living in a world shaped by heritability that it is hard for us to comprehend the case where there is no heritability.
I’ll give you examples of truly environmentally-shaped chareristics: career interests, mustical tastes, table manners, foreign language ability.
Intelligence is nothing like those in terms of variability.
If IQ were not heritable, we would see massive shifts from the schools people attemded, the teachers they had, etc. People would spend millions on their children’s education. The elite would have an army of private tutors for their children.
Blank Slate means *100% malleable*. Clay.
Not an r-squared of .67. Which is almost what height has. (Which is truly incredible given how well growth charts predict height despite not knowing anything about future diet, drug use, medical concerns, etc.)
Remember, all we believe is that IQ is *partially* heritable. We believe environment has a strong, large role.
It’s the Blank Slaters that have somehow convinced themselves that there is zero role for genes. And keep on lying to themselves ans others.
And will tell you to your face that a correlatiom of .67 is nothing. Because they haven’t worked through the implications of their beliefs.
1
3

Akin to one sibling being tall and the other one short. On the whole, height is still greatly heritable
1
1
138

Here;s a little more to chew on while I drive.
ERVs, Syncytin, and the Stacked Improbability Problem
ERVs, endogenous retroviruses, are often presented as one of the strongest arguments for common ancestry. The common explanation is that ancient retroviruses infected the germline of animals, inserted their DNA, and those viral sequences were then passed down through generations.
For years, many ERV sequences were treated as leftover junk, genetic debris from ancient infections.
But that framing has become much harder to maintain. We now know that some ERV-derived sequences are functional, regulatory, and in some cases tied to extremely important biological systems.
The clearest example is syncytin.
Syncytins are involved in placental development. They help form fused trophoblast layers at the maternal-fetal interface. In mice, syncytin-A is so important that knockout embryos die in utero. Syncytin-B knockout disrupts placental layer formation. In humans and other primates, syncytin-1 and syncytin-2 are involved in trophoblast fusion and placental function.
So this is not a trivial case of junk DNA doing something minor.
This is about reproduction, embryonic survival, and placental development, one of the most mission-critical systems in mammalian biology.
The Popular Story Sounds Simple
The usual explanation goes something like this:
A retrovirus infected an ancestor, inserted an envelope gene into the genome, and that viral gene was later co-opted for placental development.
At first, that sounds neat. Retroviral envelope proteins can mediate membrane fusion, and placentas need controlled cell fusion.
But that summary leaves out most of the actual difficulty.
Because this did not supposedly happen once.
Primates have syncytin-1 and syncytin-2. Mice and related rodents have syncytin-A and syncytin-B. Other mammal groups, including rabbits, carnivores, ruminants, and marsupials, have their own distinct syncytin-like genes.
These are generally treated as separate viral capture events, not as one inherited syncytin gene from a common mammalian ancestor.
So the real claim is not:
One viral gene entered the genome and became useful.
The real claim is closer to this:
Different retroviruses independently infected different mammalian lineages, inserted envelope genes into the germline, avoided harmful effects, escaped silencing, acquired placental expression, became regulated by the host, produced controlled cell fusion, improved reproduction, spread through populations, became fixed, and in several cases became essential to embryonic survival.
That is a very different claim.
This Is Not Just Incredulity. It Is a Stacked Probability Problem.
People often dismiss this objection as mere incredulity. But that misses the point.
The issue is not that one step sounds unlikely in isolation. The issue is that the syncytin story requires a long chain of conditional events, where each step must succeed before the next one matters.
For the viral-origin story to work, all of the following must happen:
A retrovirus must infect the germline or an early embryo in a heritable way.
The insertion must land somewhere that does not destroy fertility or development.
The viral envelope gene must remain intact enough to function.
The host must not silence or degrade it.
The gene must become expressed in the right tissue, the placenta.
It must be expressed at the right developmental stage.
Its fusogenic activity must be controlled, not chaotic or harmful.
It must coordinate with existing placental gene networks.
It must improve reproductive success enough to spread.
It must become fixed in the population.
It must eventually become integrated deeply enough that removal disrupts placental development or causes embryonic death.
That is a compounding probability problem.
Even if each individual step were assigned a modest probability, the combined probability drops quickly because the requirements are sequential and dependent.
And then the problem gets much worse: the model requires this same general chain to occur repeatedly across different mammalian lineages, involving different viral genes, but producing similar functional outcomes in placental biology.
That is the core issue.
Multiple Different Viruses, Similar Biological Outcome
This is where the syncytin case becomes especially difficult.
The standard model says primate syncytins and mouse syncytins came from different viral captures after the primate-rodent split. Other mammalian syncytins are also generally interpreted as separate captures.
So we are not being asked to accept one lucky viral insertion.
We are being asked to accept multiple independent viral insertions, from different retroviral sources, in different animal lineages, all somehow ending up tied to the same basic biological function: placental development.
That is not merely convergence. That is repeated convergence inside one of the most sensitive and indispensable systems in mammalian life.
A generic fusogenic protein is not enough. Placental development does not merely need fusion. It needs fusion in the right cells, in the right tissue, at the right time, under tight regulatory control, without killing the embryo, damaging the mother, wrecking maternal-fetal exchange, or triggering the wrong immune response.
A viral envelope protein can fuse membranes. Fine.
But a placental system requires controlled, coordinated, developmentally integrated fusion.
Those are not the same thing.
The Mission-Critical Problem
This matters because the placenta is not an optional feature.
In placental mammals, proper placental development is necessary for embryonic survival. If the system fails, the organism does not reproduce. In mice, disruption of syncytin-A is embryonic lethal. That means the proposed viral acquisition did not merely add a side benefit. It allegedly became part of a system without which the organism cannot continue.
That creates a serious explanatory problem.
If the placental system required syncytin-like function, then how did it operate before that function existed?
If it did not require syncytin-like function, then how did a viral insertion become so deeply integrated that later removal breaks the system?
Either way, the model has to explain not just origin, but transition.
It has to explain how a reproductive system survived before the viral gene, how the viral gene entered without damaging that system, and how the gene later became essential.
That is not a small detail. That is the heart of the problem.
Fixation Makes the Problem Harder
It is also not enough for a viral insertion to happen in one animal.
For the syncytin story to work, the insertion must become heritable and then fixed in a population. That means it must spread until it is carried broadly across the lineage.
Modern humans do show some polymorphic HERV-K insertions, meaning certain ERV-like insertions are present in some people and absent in others. But that is not the same thing as directly observing a new viral envelope gene become fixed in the entire human population because it generated an essential developmental function.
That distinction matters.
The syncytin model depends on rare insertions becoming inherited, useful, regulated, fixed, and eventually indispensable.
One such event would already require explanation.
Several such events, independently producing similar placental functions, require much more.
The Missing Parent Virus Problem
There is also no exact ancestral virus sitting in a freezer that can be compared directly to syncytin-1, syncytin-A, syncytin-B, syncytin-Car1, syncytin-Rum1, or the others.
The viral-origin claim is inferred from sequence similarity, retroviral envelope-like structure, LTRs, and phylogenetic analysis.
That evidence may show that syncytins resemble retroviral envelope genes. But resemblance is not the same as demonstrating the full causal pathway.
It does not directly show the infection event.
It does not show the intermediate stages.
It does not show how the host survived the transition.
It does not show how the gene became properly regulated.
It does not show how separate events repeatedly produced similar placental outcomes.
So the real burden is much larger than simply saying these genes look viral.
The real burden is explaining how viral-looking genes became mission-critical components of mammalian reproduction, multiple times.
Why the Junk-DNA Story Should Make People More Cautious
The syncytin case should also make people cautious about the old junk-DNA assumptions.
Sequences once dismissed as viral leftovers are now known to be involved in major biological functions. Some are regulatory. Some are tissue-specific. Some are tied to development. Syncytin is not just active; it is involved in reproduction and embryonic survival.
That should at least raise a serious question:
Were these sequences really accidental viral debris later patched into useful roles, or are we looking at functional genomic components that were too quickly interpreted through an evolutionary lens?
The standard answer assumes viral origin first and then explains function later.
But the more function we discover, especially in critical systems, the less satisfying the ancient viral accident framing becomes.
The Better Explanation: Common Design
From a design perspective, the pattern is not surprising.
Different mammalian groups need to solve similar biological problems: controlled cell fusion, maternal-fetal exchange, immune modulation, trophoblast-layer formation, and placental development.
It makes sense that similar functional modules would appear in different mammalian systems.
The standard viral-origin model says this pattern was produced by multiple independent viral accidents, filtered by selection, and gradually integrated into reproduction.
The design model says similar biological problems are solved with similar engineered tools.
The first explanation depends on stacked, repeated, historically unobserved events producing mission-critical outcomes.
The second explanation treats the pattern as intentional reuse of functional design principles.
At minimum, syncytin should not be presented as a simple victory for the viral-origin story. It is far more complicated than that.
The evidence may show viral-like features.
But the explanation still has to account for the compounding improbability of multiple independent infections, heritable germline insertions, beneficial regulation, population fixation, and repeated integration into one of the most essential systems in mammalian biology.
That is the syncytin problem.
It is not just that it sounds unlikely.
It is a legitimate cumulative-probability problem hiding inside one of the most popular ERV arguments.
References for Further Reading
Mi et al., 2000: identification of syncytin-1 as a retroviral-envelope-derived gene involved in human placental morphogenesis.
Dupressoir et al., 2005: identification of syncytin-A and syncytin-B in mice as placenta-specific fusogenic genes.
Dupressoir et al., 2009: syncytin-A knockout causes embryonic death and placental defects.
Dupressoir et al., 2011: syncytin-B knockout disrupts syncytiotrophoblast layer formation.
Cornelis et al., 2015: review of retroviral envelope gene captures and syncytin exaptation across mammals, including marsupials.
Vernochet et al., 2014: review of mouse syncytin-A/B and multiple independent syncytin captures in mammalian lineages.
Grandi and Tramontano, 2018: overview of HERVs as roughly 8% of the human genome and discussion of HERV domestication.
Kahyo et al., 2017; Belshaw et al., 2005: HERV-K insertional polymorphism in humans.
2
1
40

What don't you understand about "EIGHTY PERCENT HERITABLE"!!! ???
Good Lord, you people are slow-witted!
There are countless "twin studies" that looked at what you just said, and REGARDLESS of environment, IQ is 80% PRE-DETERMINED by genes!
9

Evolution:
The change in the heritable characteristics of biological populations over successive generations.
That's the definition that underpins all of biological, medical, paleontological, science. And many many other fields of science. It's in all textbooks Ari and the world from highschools to universities. (Or some derivative of it mentions specifically the allele frequencies)

2

How should this be possible ? I’m assured by real genetics “HBD” scholars there are Studies that show political opinion is heritable and therefore the right’s long term victory is assured by differences in birth rates.
Jun 14

I think it’s funny people never addresses Charlie Kirk younger sister who has completely different political beliefs and is huge Bernie Sanders fan who supporting Palestine


45
29
791
33,408

“Rich and successful parents have smart kids, intelligence is highly heritable, intelligence is a good predictor of future social standing, and things that correspond with being stupid also correspond with being poor, but I would never ever connect the dots between any of this.”
5
96


BIOLOGY / BLACK CRIME
Group differences in traits like intelligence, impulse control, and aggression have a substantial genetic component, and these feed directly into the violence patterns.
IQ AND CRIME LINK
Average Black American IQ is ~85 (vs. ~100 for Whites, ~106 for East Asians). This ~15-point gap has held for decades in massive datasets (SAT, military, NAEP, etc.).
IQ is ~50-80% heritable within populations (twin/adoption studies; rises with age).
The gap predicts real outcomes: lower IQ strongly correlates with higher crime, impulsivity, poor future-time orientation, and welfare dependency.
Low cognitive ability limits impulse control and opportunity costs for crime.
The violence isn’t evenly spread across “the 13-14%.”
It’s hyper-concentrated in young Black males (~3-7% of total US pop), where arrest rates hit ~49% by age 23 (vs. 38% White males).
A small chronic subset drives 50% of murders/robberies.
Biology amplifies this: genes influence brain development, executive function, and testosterone responses.
GENETIC EVIDENCE
• Heritability doesn’t vanish between groups: High within-group heritability persistent gaps after SES controls regression to racial means in adoption/transracial studies (Minnesota study: Black adoptees in White homes still lag) point to partial genetic causation.
Admixture studies (lighter skin/higher European ancestry = higher IQ) and polygenic scores align.
Environmental explanations (poverty, racism, nutrition) fail to close the gap despite trillions spent and Flynn effects that don’t equalize races.
1
12

Heritable mutation that sometimes produces things of value but is otherwise a defect. Rise of Autism has been connected to rise of synthetics more chemicals in food and cooking ect. Some autistic people would’ve always been around but rising numbers concerning.


2
7
245

with the awareness that some men were always going to become rapists regardless of socialization comes the awareness that some men would never even *think* about raping a woman, regardless of socialization or opportunity... and these behavioural traits are *heritable*.
1
7
154

When they first started doing selection experiments on Drosophila, they found that virtually every trait could be selected. Even the tendency to walk right rather than left. Because all was heritable. Sasha thinks that human intelligence isn't heritable. Evolution deniers.
1
51

Yes, three very different situations:
1. Everyone gets IQ 115 forever.
2. Everyone gets IQ 115, and it's 80% heritable from there.
3. Everyone gets IQ 115, but it's back to normal for the next generation.
2
4
73