Hang Zheng retweeted

May 7
Tired of seeing the same binding-affinity-prediction benchmarks time and time again?
The OpenBind initiative just released a big bolus of data (xtals affinity), and we used a curated subset of this dataset to test Rowan's structure-prediction and RBFE workflows.
1
12
112
14,212

Jun 10
Already true for protein. IsoDDE only available w royalty and upfront prohibitive to small biotech, but we are the ones who discover most drugs! This was inevitable without orgs like @open_fold which beat AF3 in openbind blind tests as well
4
1,009
FWIW if you look at ligand in openbind truly blind testing OF3 is beating AF3 there (Protenix too, but w later training cutoff aka more data)
1
551

Jun 4
All the people doing protein interaction predictions. With the release of openBind, why not train a model on that, then fine-tune it on PPI?

4
4,888

in our hands, Pearl is the strongest system for protein-ligand structure prediction. we see it daily in live drug discovery programs. now SOTA on OpenBind, surpassing every cofolding model in the zero-shot regime. proud of this work. more to come!

Today we're sharing new breakthrough results for Pearl, our foundation model for protein–ligand cofolding.
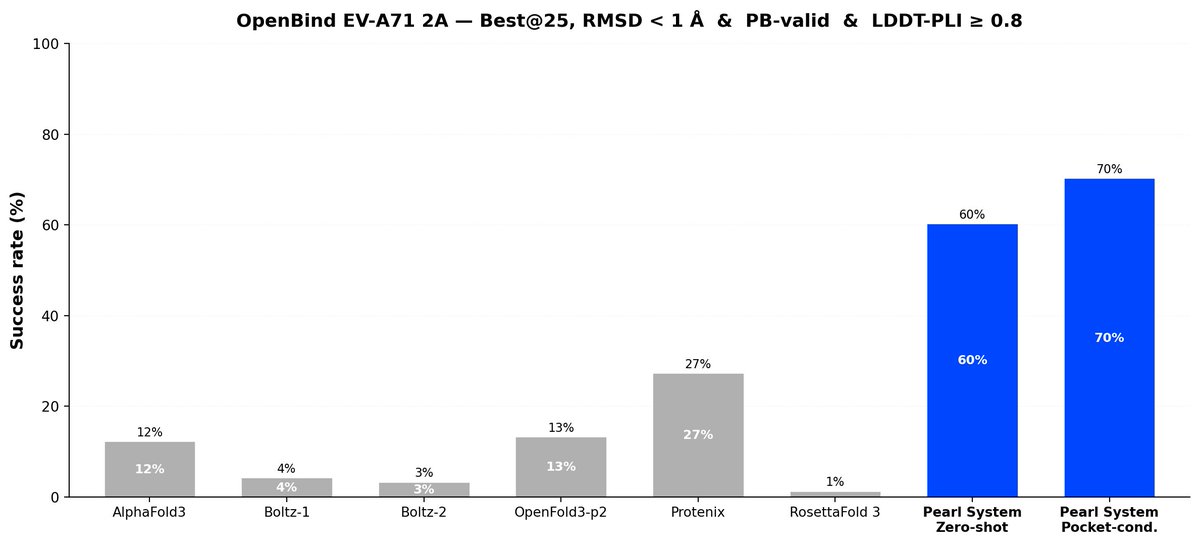
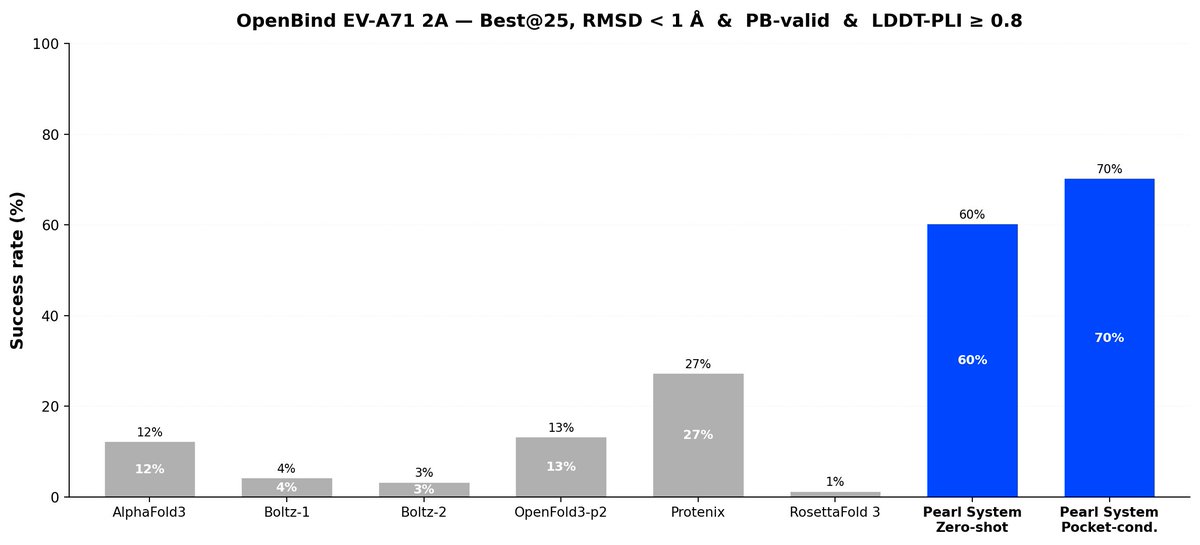
The OpenBind Consortium recently released the first public structure-affinity benchmark for molecular AI, evaluating six prominent cofolding models on the EV-A71 2A protease. We ran our full Pearl system against the same target.
Zero-shot, with no binding-site information and no tuning, the Pearl system reaches 78% on OpenBind's primary success criteria, far ahead of every cofolding model tested by OpenBind. We also assessed a stricter sub-1 Å accuracy threshold, which is more relevant for real-world R&D usage – the Pearl system’s success is still 60%, versus 1–27% for the other models.
What matters most to us: this is the same system setup our scientists use on live drug discovery programs, not a benchmark-specific configuration.
Thanks to the OpenBind Consortium for building a rigorous public benchmark, and to @NVIDIAHealth for the support on optimizations that enabled model scaling.
4
140

Jun 3
We ran Pearl on OpenBind, a benchmark created to challenge co-folding and assess the field. It surpassed all other models — zero-shot.
Our Pearl system exceeded the other models on every metric, particularly on the most relevant measures that correspond to performance on real-world drug programs.
More detail for fellow nerds👇
genesis.ml/news/zero-shot-pe…
1/ Zero-shot means zero added help.
Pearl got a protein sequence, a ligand SMILES, and an unbound template. Nothing else. (We also tested it providing a few binding-site residues, but zero-shot performance was already outstanding.
→ 78% on OpenBind's triple success criteria. Closest next best is 54%.
2/ 2Å is too fuzzy. The number that matters: sub-1Å.
That's the real RMSD accuracy threshold that’s relevant to be useful in practice (for actually designing molecules and downstream predictions like potency).
→ Pearl: 60% → Best competitor: 27% → Most models: 1–13%
At this bar, Pearl beats every method — including classical docking that gets the bound crystal structure Pearl never sees.
3/ This target is hard.
A loop at the binding site shifts ~4Å when a ligand binds. The pocket doesn't even fit a ligand until the protein moves. Classical docking from the unbound structure can’t model this.
This also isn’t a target with similar structures in the PDB, so co-folding models can’t cheat. Unlike some benchmarks, this makes the OpenBind challenge a proper test of real-world utility.
Pearl predicts the motion from sequence alone and handles the new system with ease – including one compound placed to 0.28Å that no other zero-shot method solved.
4/ How we did it.
Synthetic data at training. Equivariant architecture. Novel inference time scaling. AI physics scoring to converge the outputs.
This is what a foundation model that generalizes and works in actual drug discovery programs looks like — not one that memorizes the PDB.
Full write-up figures 👇
2
7
48
5,125

Jun 3
A month ago, OpenBind released their first public structure-affinity benchmark. Our team ran the Pearl system on it, zero-shot, with no tuning for the target, and it came out ahead of every cofolding model evaluated, especially at the sub-angstrom accuracy that actually matters for drug discovery. Genuinely proud of our team here.
genesis.ml/news/zero-shot-pe…
6
10
43
81,478

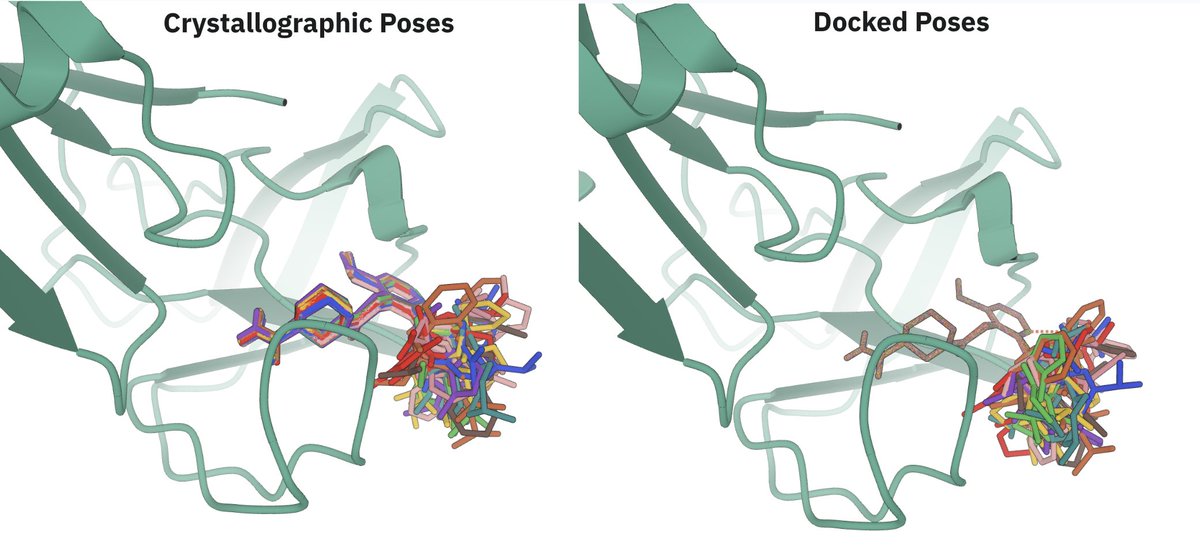
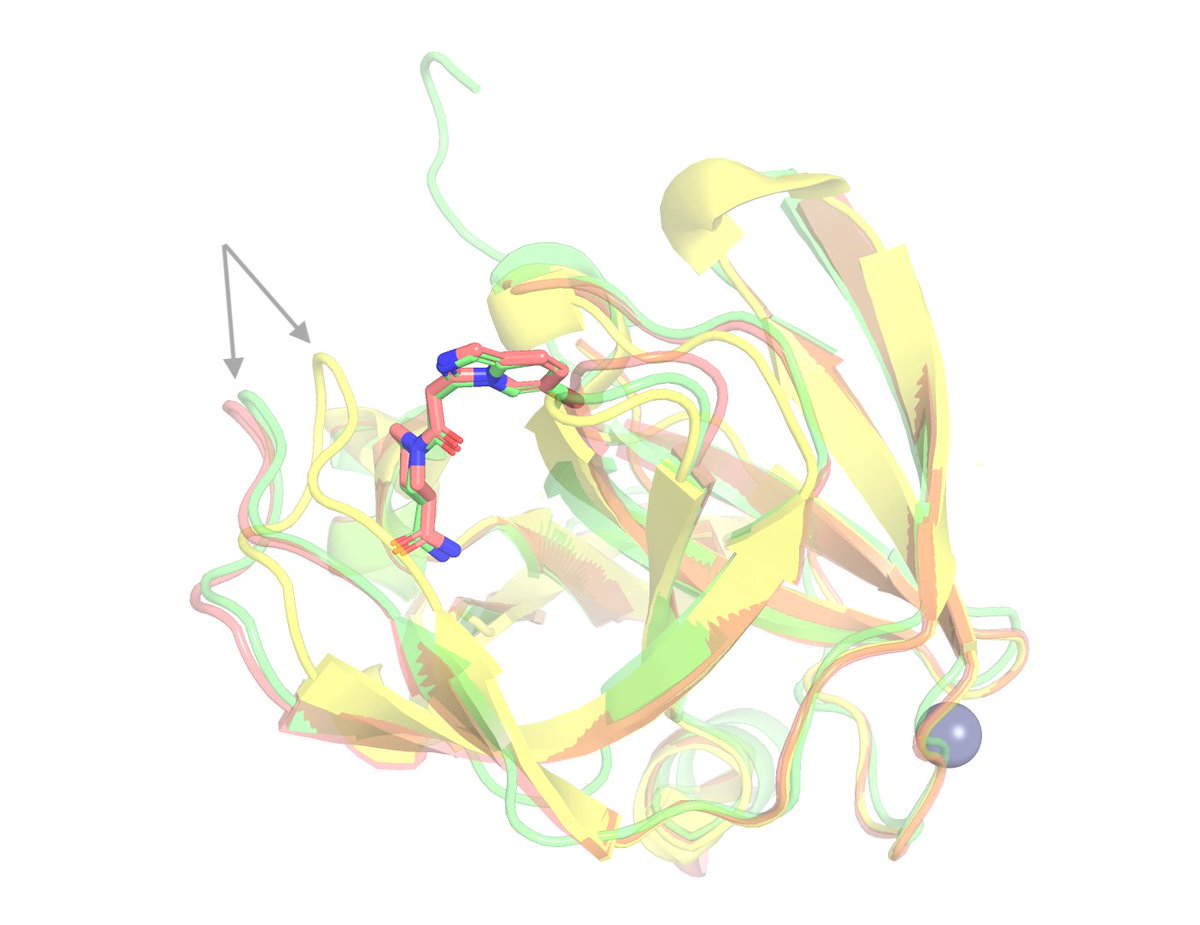
Why is the EV-A71 2A protease such a hard target?
When a ligand binds, a loop next to the binding site shifts by roughly 4 Å (as shown in the image below). Every one of the 802 structures in OpenBind's benchmark requires that rearrangement, which is why classical docking into the unbound structure has no practical utility (under 5% success).
Success requires predicting the protein moving, from sequence and the ligand alone. Co-folding is capable of this, but the target is particularly challenging given its dissimilarity to training data in the Protein Data Bank. Most benchmarks struggle to test ability to generalize, so we applaud OpenBind for releasing a complex system as a robust test case.
The Pearl system is uniquely capable of generalizing to this target, capturing this induced fit, and achieving high predictive performance. We accomplished this by integrating novel inference time scaling methods along with physics- and AI-based pose ranking, which is a big part of why Pearl holds up on real programs and not just benchmarks.
1
1
7
364
Today we're sharing new breakthrough results for Pearl, our foundation model for protein–ligand cofolding.
The OpenBind Consortium recently released the first public structure-affinity benchmark for molecular AI, evaluating six prominent cofolding models on the EV-A71 2A protease. We ran our full Pearl system against the same target.
Zero-shot, with no binding-site information and no tuning, the Pearl system reaches 78% on OpenBind's primary success criteria, far ahead of every cofolding model tested by OpenBind. We also assessed a stricter sub-1 Å accuracy threshold, which is more relevant for real-world R&D usage – the Pearl system’s success is still 60%, versus 1–27% for the other models.
What matters most to us: this is the same system setup our scientists use on live drug discovery programs, not a benchmark-specific configuration.
Thanks to the OpenBind Consortium for building a rigorous public benchmark, and to @NVIDIAHealth for the support on optimizations that enabled model scaling.
1
22
52
6,463

Building the Next AlphaFold for Drug Discovery
@IsomorphicLabs closed a $2.1 billion Series B to commercialize AlphaFold — but Charlotte Deane's argument in this clip is that the next equivalent breakthrough is still waiting on the data infrastructure to make it possible.
Deane makes a point that tends to get lost in the excitement around model capabilities: AlphaFold succeeded in large part because the Protein Data Bank existed, an accumulated fifty years of experimental results that gave AI something substantive to learn from. Her @openBIND consortium is an attempt to replicate that condition deliberately — building the dataset for protein-ligand binding with AI requirements built in from the start, not discovered after the fact. The $2.1 billion now flowing into AI drug discovery, as reported by @BuildFastWithAI, will accelerate what is already possible with existing data, but Deane's work is about removing the ceiling on what comes next.
For institutional decision-makers evaluating where durable value is being created in this space, the more consequential investment question may not be which model is best today, but who is building the data infrastructure that determines what models can do in five years.
We've put a link to the full episode with Charlotte Deane and our hosts @stephenjhorn and @Laila_Rizvi in the first comment.
#BiomedicalDataScience #AIMedicine #ComputationalBiology #AlphaFold #AIDrugDiscovery
1
2
58
May 20
As promised, we've released a followup post exploring the OpenBind data in more detail:
- how we modified settings compounds to find useful accuracy
- what's still going wrong
- if these results would actually be useful
- how we plan to fix systems like this in the future.
May 7

Tired of seeing the same binding-affinity-prediction benchmarks time and time again?
The OpenBind initiative just released a big bolus of data (xtals affinity), and we used a curated subset of this dataset to test Rowan's structure-prediction and RBFE workflows.
2
6
58
9,635

May 19
#Innovadora | 💊El proyecto #OpenBind acerca la IA a una nueva era en el diseño de #medicamentos
➡️ La publicación de datos experimentales del consorcio británico marca un hito en los esfuerzos por mejorar el uso de la #IA en el descubrimiento de nuevos #fármacos
consalud.es/industria/innova…
1
142

5/17 10AMからPaper Talkを配信します
今回は5月前半にポストした研究に関してLLMで作った概要図を使って紹介していきたいと思います
【#PaperTalk #9】 バイオインフォマティクス最新研究紹介 2026年5月前半 mTM-align2 OpenBind Genie3 ... youtube.com/live/gayaKrFlluU…
2
7
3,256

May 14
The OpenBind consortium’s first release of experimental data marks a milestone in efforts to improve how artificial intelligence (AI) is used in drug discovery.
Find out more ⬇️
ox.ac.uk/news/2026-05-12-ope…

ALT The image shows a colourful abstract scientific visualisation featuring ribbon-like protein structures, molecular models and translucent flowing forms in bright shades of orange, blue, yellow and pink. Overlaid text reads: “OpenBind releases first open dataset and AI model for drug discovery” alongside University of Oxford branding.
3
11
64
14,828
May 14
Of course OF3p2 is the download now on the openfold3 github. Openbind results are fascinating, it beats AF3 on that set of new xtals!
2
42

L'initiative OpenBind publie son premier jeu de données IA pour accélérer la découverte de médicaments 💊 Les détails ⤵️ #PharmaTech #IA enerzine.com/openbind-marque…
1
2
73

May 7
Large, shared datasets have repeatedly become engines of scientific progress. The Protein Data Bank helped make modern structural biology much more learnable. UK Biobank and deCODE helped transform population-scale human genetics.
You can see the same pattern in newer efforts to build AI-enabling datasets, such as OpenBind, EvE bio, and OpenADMET. They span binding, broad drug-target pharmacology, and ADMET properties, but share the same basic bet that better data will make better biological AI possible.
The teams deciding what to measure, curate, and share now are shaping what biology will be able to do with AI in the next few years. They are expanding what future biology will be able to discover.
1
2
174
EV-A71 2A proteaseを対象にKD値を紐付けたstructure-affinity dataset、OpenBindが公開されました。ベンチマークでは受容体の形状変化(conformational mismatch)がdocking精度の主因として可視化され、affinity予測は分子量という単純指標にも及ばない現状が明らかになりました。
ソースはリプ🔽
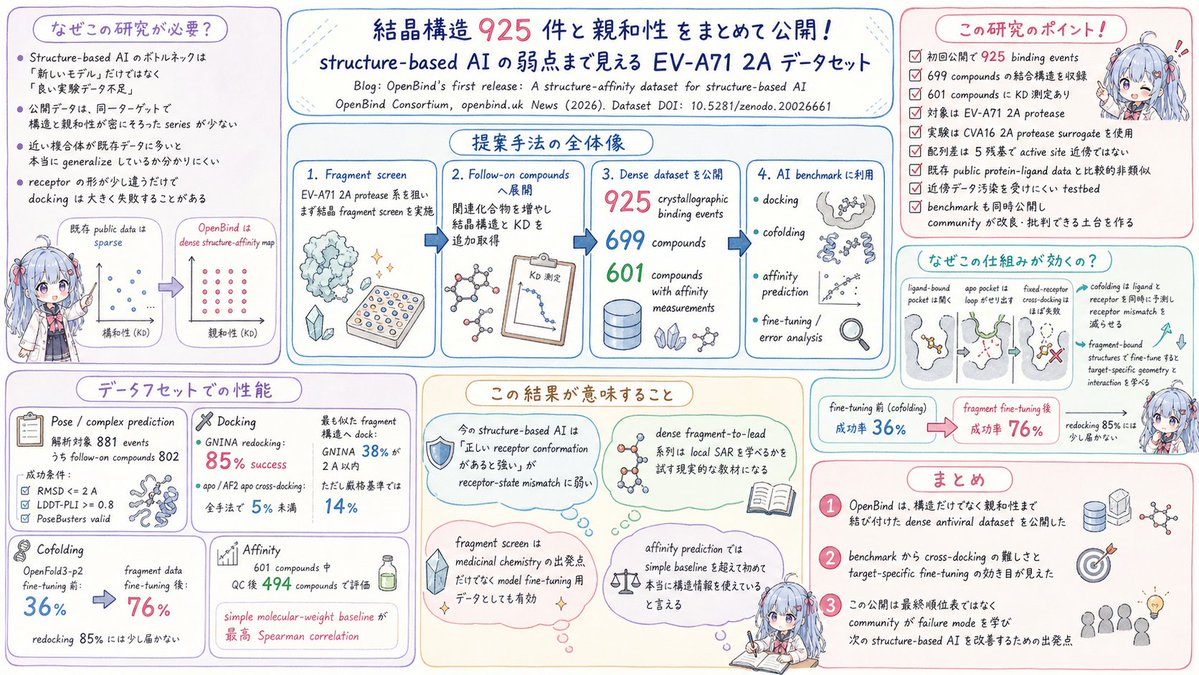
ALT 概要図は ChatGPTを用いて作成しています。 内容の理解を助けるための要約図のため、構造式、細かな数値や表現、ニュアンスに一部誤りが含まれる可能性があります。 特には構造式は大きく構造が異なる可能性が高いので、あくまでもLLMによる模式図と捉えて下さい。
1
4
13
1,342
Excited to have @open_fold collaborate with OpenBind on model training, results so far look very exciting!

NEW: today OpenBind ‘comes out of stealth’ so to speak with their first data dump of ~900 novel protein-ligand structures - most with paired affinities
This represents a meaningful %-age increase in all of humanities P-L data in the PDB collected in the last 50 years
More👇

1
1
3
588
May 7
Good benchmarks drive scientific advancement, and we're very excited to continue learning from the OpenBind data sharing what we learn. We encourage others to try this new dataset out with their methods!
Check out the full blog here:
rowansci.com/blog/testing-ro…
11
696