
Make sure you get the relocalization mod. It's still localized compared to the JP original, but it's far less sloppy.
7

Google AI :
"Demand for Remote Work: Soaring travel expenses have caused a massive spike in employees requesting flexible hours or full remote work.Job Relocalization: Rising fuel costs are forcing workers to
1/
1
133

Jun 14
wait theres a relocalization for nier??? ive been wanting to play the series but didnt wanna play the remake cause it looks like dogshit
2
1
217

Jun 9
This was unexpected from Gemini
Today was a surprise; I was comparing approaches to a relocalization problem using Gemini Pro 3.1 and it automatically created this simple yet intuitive console for me to test out the long-term drift in each approach.
1
28

Jun 9
...Also NSP2 recruits the GIGYF2 to viral double-membrane vesicles (DMVs), where viral RNA replication and translation occur. This relocalization repurposes a key quality-control machinery, diverting it from its normal cellular function in ribosome-associated quality control...
1
15

2/ The numbers:
→ 150,000 sqm mapped
→ 230 shops
→ Multi-floor, high-traffic environment
→ Sub-100ms relocalization latency
GPS dies the moment you walk indoors. Our VPS doesn't.
1
1
16
861

May 30
The formal version of the Information Theory of Aging was first formulated in 2014 while I was in Fiji and was later described in my book Lifespan: Why We Age—and Why We Don’t Have To Die.
The idea started in the 1990s, when I was working in the laboratory of Leonard Guarente at MIT, one of the pioneers of modern aging research.
At a time when many scientists viewed aging as an inevitable process of wear and tear, Lenny’s laboratory was helping reveal that lifespan could be regulated by specific genes and cellular pathways.
The groundbreaking work on the SIR genes in yeast (which stands for silent information regulator) established a connection between chromatin structure, gene regulation, and longevity that continues to influence the field today.
As part of that research team, I became interested in a simple but profound paradox. If most cells maintain essentially the same DNA sequence throughout life, why do they progressively lose function? Why do young cells know how to repair themselves and maintain their identity, while old cells gradually lose those abilities?
Even in yeast, it was clear that aging cells were losing control of gene expression despite retaining much of their genetic information. These observations led me to wonder whether aging might involve more than the accumulation of mutations and molecular damage. Perhaps cells were losing access to some of the information required to read the genome correctly. This was the theory’s origins in 1995-1999
Over the following years, this idea continued to evolve. In my laboratory at Harvard Medical School, Philipp Oberdoerffer led a series of experiments examining how chromatin regulators respond to DNA damage. In a landmark study (Oberdoerffer et al., Cell, 2008), we found that proteins involved in maintaining chromatin structure relocate across the genome to help repair DNA breaks. While this response protects the genome, it can also alter patterns of gene regulation. We proposed that repeated cycles of DNA repair might gradually disrupt epigenetic organization over time, generating biological noise that accumulates throughout life. This became known as the Relocalization of Chromatin Modifiers hypothesis and provided an important conceptual foundation for later work.
For many years, however, these ideas remained largely speculative. Then a series of discoveries began to provide support.
One came from Steve Horvath’s development of epigenetic clocks (Horvath, Genome Biology, 2013). By measuring DNA methylation patterns, Horvath showed that biological age could be predicted with remarkable accuracy across many tissues. For the first time, aging appeared to leave a measurable signature on the epigenome itself.
Another important advance came from the laboratory of Juan Carlos Izpisua Belmonte. In 2016, his group demonstrated that transient expression of the Yamanaka factors could reverse multiple features of aging and extend lifespan in a mouse model of progeria (Ocampo et al., Cell, 2016). The work suggested that aspects of biological age might be reversible without permanently erasing cellular identity.
Around 2010, my laboratory began designing experiments to directly test whether disruption of epigenetic information could itself drive aging in a mammal, not just a yeast cell…
4
11
108
7,265






May 9
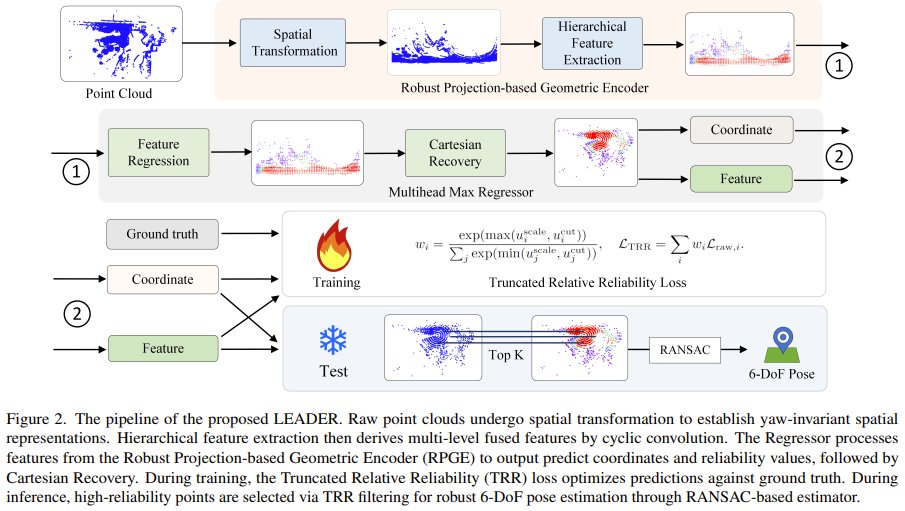
Can your robot always know exactly where it is, even in noisy, complex environments?
Researchers from Xiamen University and University of Bristol present LEADER — a new LiDAR relocalization method that doesn't treat all points equally.
Instead, it uses a smart geometric encoder to capture multi-scale shapes and a “reliability loss” that ignores unreliable predictions, reducing the impact of noise.
Result: Outperforms state-of-the-art methods by 24.1% lower position error on the Oxford RobotCar dataset and 73.9% lower on the NCLT dataset. Code released.
LEADER: Learning Reliable Local-to-Global Correspondences for LiDAR Relocalization
Paper: arxiv.org/abs/2604.11355
Repo: github.com/JiansW/LEADER
Our report: mp.weixin.qq.com/s/Swkvg5mRN…
📬 #PapersAccepted by Jiqizhixin
3
19
1,897


Apr 26
I heard that after reading your wise posts Ryukishi himself commissioned Dragonbaby to do a full relocalization of Umineko in the Yunyun style. Sounds great, doesn't it? Let's get hyped for 'good translation' gems like 'Without love, you can't see the fascists!' to own the chuds!
1
1
45
3,257

Apr 23
𝗧𝗥𝗔𝗡𝗦𝗩𝗜𝗦𝗧𝗔'𝘀 𝗠𝗶𝗻𝗶𝗮𝘁𝘂𝗿𝗶𝘇𝗲𝗱 𝗧𝘄𝗼-𝗣𝗵𝗼𝘁𝗼𝗻 𝗧𝗲𝗰𝗵𝗻𝗼𝗹𝗼𝗴𝘆 𝗛𝗲𝗹𝗽𝘀 𝗥𝗲𝘃𝗲𝗮𝗹 𝘁𝗵𝗲 𝗡𝗲𝘄 𝗠𝗲𝗰𝗵𝗮𝗻𝗶𝘀𝗺 𝗼𝗳 𝗔𝘀𝘁𝗿𝗼𝗰𝘆𝘁𝗲-𝗥𝗲𝗴𝘂𝗹𝗮𝘁𝗲𝗱 𝗚𝗹𝘆𝗺𝗽𝗵𝗮𝘁𝗶𝗰 𝗙𝘂𝗻𝗰𝘁𝗶𝗼𝗻 𝗶𝗻 𝗔𝗹𝘇𝗵𝗲𝗶𝗺𝗲𝗿'𝘀 𝗗𝗶𝘀𝗲𝗮𝘀𝗲
Alzheimer's disease is a progressive neurodegenerative disorder that starts decades before cognitive symptoms appear, with amyloid beta and tau tangles accumulating in the brain. As treatments advance, effective biomarkers are needed for different disease stages. Recent studies highlight the glymphatic system's key role in clearing brain pathologies, but more cellular-level research is required.
We would like to acknowledge an interesting research study published in 𝗡𝗮𝘁𝘂𝗿𝗲 𝗡𝗲𝘂𝗿𝗼𝘀𝗰𝗶𝗲𝗻𝗰𝗲, which investigates abnormal calcium dynamics in astrocytes during the early events of Alzheimer's disease and its impact on the glymphatic system. Congratulations to Drs. Yamei Tang, Wei-Jye Lin, Xiaojing Ye and their teams from Sun Yat-sen University for this outstanding work on Alzheimer's disease.
𝗪𝗵𝗮𝘁'𝘀 𝗜𝗻𝗻𝗼𝘃𝗮𝘁𝗶𝘃𝗲:
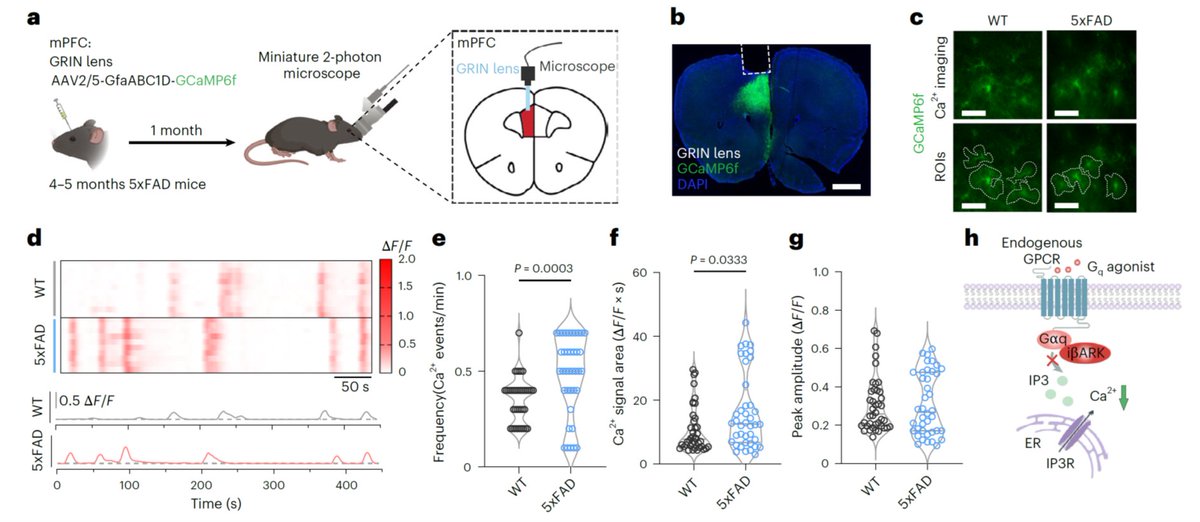
1️⃣ Abnormal Calcium Dynamics in Astrocytes: The study demonstrates a functional connection between amyloid-β-induced elevation of calcium dynamics in medial prefrontal cortex (mPFC) astrocytes and glymphatic dysfunction in cognitively impaired 5xFAD mice.
2️⃣ Disruption in AQP4 Polarity: Calcium hyperactivity increases cholesterol synthesis in astrocytes, which leads to increased endocytosis of aquaporin-4 (AQP4) and its relocalization to lysosomes.
3️⃣ Rescue of Cognitive Decline: Selective suppression of Gq GPCR-mediated calcium activity in astrocytes (achieved both in vitro and in vivo) significantly improved deficits across multiple cognitive domains previously linked to the mPFC. Notably, anxiety-like behaviors or general locomotor activity were not affected.
4️⃣ Restoration of Glymphatic Reflux: Suppressing Aβ-induced hyperactivity of astrocytic calcium restored glymphatic influx and reversed the disrupted AQP4 polarity.
5️⃣ Cholesterol Biosynthesis: Transcriptional profiling reveals cholesterol biosynthesis as a downstream pathway regulated by astrocytic calcium activity.
𝗠𝘂𝗹𝘁𝗶𝗽𝗵𝗼𝘁𝗼𝗻 𝗜𝗺𝗮𝗴𝗶𝗻𝗴 𝗶𝗻 𝗙𝗿𝗲𝗲𝗹𝘆 𝗕𝗲𝗵𝗮𝘃𝗶𝗻𝗴 𝗠𝗶𝗰𝗲: The study presented exemplary findings in calcium dynamics in the mPFC of 5xFAD mice, utilizing a GRIN lens for enhanced imaging.
At TRANSVISTA (transvista.com), we are committed to supporting cutting-edge neuroscience by providing tools that expand what is possible in brain and behavior research.
Once again, congratulations to the authors and research teams for their work in Alzheimer's disease.
Read Article Here: doi.org/10.1038/s41593-026-0…
#Alzheimer's #TwoPhotonImaging #CalciumImaging #FreelyMovingMice #BrainResearch #ScientificData #Neuroscience

3
3
289

Apr 21
Countries such as Russia and France must account for reputational risk when entering technology transfer or relocalization agreements with India, particularly after India has proved to be a complete PR disaster for their flagship military equipment (Su-30 and Rafale).
4
4
13,302

Apr 17
"A Scene is Worth a Thousand Features: Feed-Forward Camera Localization from a Collection of Image Features"
TL;DR: feed-forward localization builds a lightweight feature map and estimates camera pose in one pass, achieving fast and accurate relocalization across large scenes
1
18
259
17,294

Apr 15
SIB1-SEC23A undergo ER to chloroplast relocalization to mediate immunity in Arabidopsis thaliana
onlinelibrary.wiley.com/doi/…
1
6
316

4/ What can the model do? Predict PPI’s, resolve compartment specific functions, predict drug-induced relocalization, predict cell states without explicit markers, enable unsupervised segmentation of subcellular compartments, and spatial decomposition of gene sets
1
3
811

No doubt massive regulatory/safety hurdles, but many aspirational therapeutic mechanisms currently bottlenecked at chemistry - cell type specific TF activation, dissolving aggregates in neurodegen. diseases, protein relocalization, among others
2
90

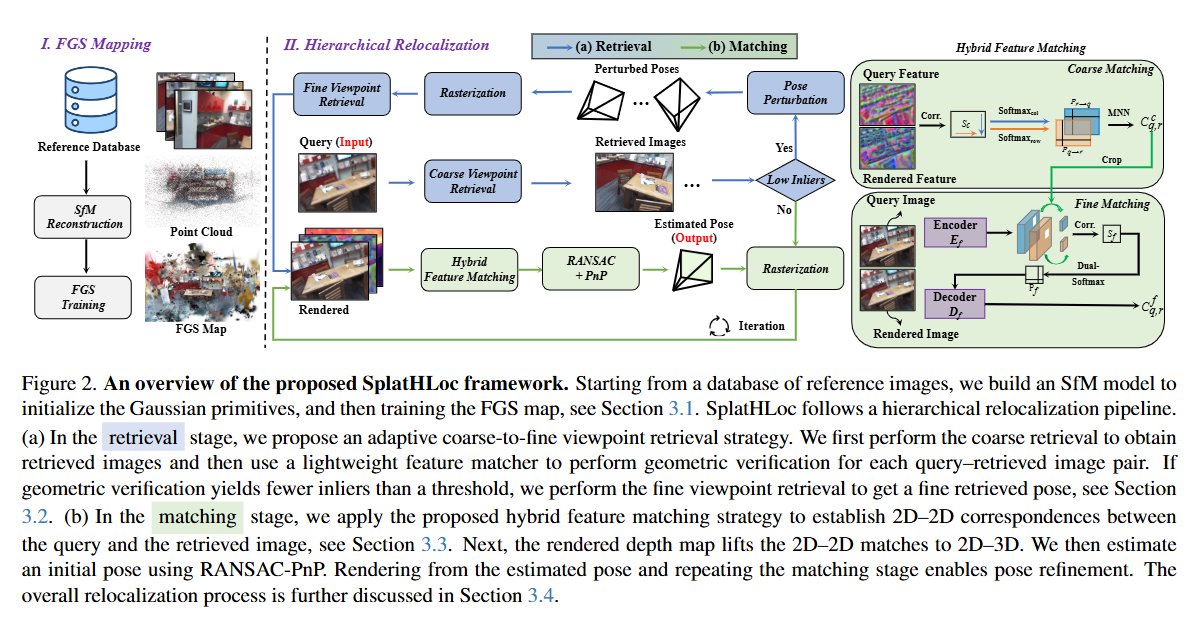
Hierarchical Visual Relocalization with Nearest View Synthesis from Feature Gaussian Splatting
Huaqi Tao, Bingxi Liu, Guangcheng Chen, Fulin Tang, Li He, Hong Zhang
tl;dr: iterative retrieval-verification-rendering; rendered feature JamMa->matching
arxiv.org/abs/2603.29185


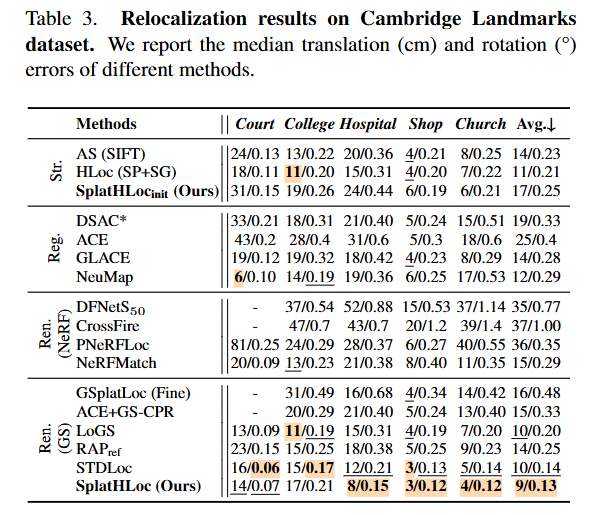
1
5
25
1,495

From @grok - happy hunting!
Here are the **most disqualifying** weaknesses, gaps, and holes that render the 2023 Cell ICE mice study (and associated Sinclair/OSK claims) largely **bunk** as proof of epigenetic information loss as a primary, causal, and safely reversible driver of mammalian aging:
- **No lifespan extension in the core ICE study** — The paper demonstrates accelerated aging phenotypes and partial molecular reversal but reports zero data on increased median or maximum lifespan in ICE mice after OSK treatment (or even in untreated controls). Claims of "reversing aging" collapse without this gold-standard endpoint.
- **Complete absence of teratoma, dysplasia, or cancer monitoring** — The word "teratoma" does not appear once in the paper despite well-documented oncogenic risks from in vivo OSK/OSKM reprogramming in prior studies; no systematic histology, tumor screening, or long-term safety data were provided.
- **Artificial induction does not mimic natural aging** — Repeated, targeted I-PpoI-induced DSBs in young mice create non-physiological, synchronized damage unlike the gradual, stochastic, diverse insults of normal aging; rDNA locus targeting introduces unruled-out confounds (rDNA instability affects lifespan in model organisms).
- **Whole-body, non-cell-autonomous design** — Systemic ICE activation prevents distinguishing direct cellular effects from secondary systemic responses; no tissue-specific controls.
- **Bulk-level data only; no single-cell resolution** — Epigenomic and transcriptomic analyses are bulk, masking cell-type heterogeneity, rare cell contributions, or variable reprogramming efficiency.
- **Mechanistic black box** — No identification of which specific chromatin factors relocalize, no in vivo chromatin contact mapping (Hi-C/HiChIP limited or absent for key claims), leaving the core "information loss via relocalization" hypothesis unproven at molecular detail.
- **Reversal is narrow, partial, and surrogate-heavy** — Phenotypic improvements limited to select tissues (retina, some kidney/muscle markers, neurons); relies heavily on epigenetic clocks and gene expression as proxies rather than comprehensive functional restoration across organs or organismal healthspan.
- **Ignores non-cellular aging components** — Extracellular matrix (e.g., irreversible collagen cross-links via glycation), persistent senescent cell effects, proteostasis decline beyond epigenetics, and other hallmarks remain unaddressed and unlikely fixed by transient OSK alone.
- **Overhyped translation to "age reversal"** — Media and author framing ("rebooting," "permanent reset," "aging forwards and backwards at will") far exceeds the data; independent replication with rigorous lifespan/safety endpoints is absent, and critics note the work leans more promotional than definitive.
These flaws mean the study provides interesting correlative or provocative data at best but fails as causal proof or a foundation for human "information theory of aging" therapies. Stronger, multifactorial models of aging (damage accumulation across compartments) remain intact. Independent, long-term studies with full organismal endpoints are essential before any claims graduate from hype.
2
2
751