
🔬Diving deep into #MECFS, #LongCOVID, #LongEBV & post-vaccine syndrome research. Dual role:Researcher and EBV ME/CFS patient. Seeking answers. #Antivirals #HLA
Joined August 2021
- Tweets 2,307
- Following 221
- Followers 5,584
- Likes 2,232
284 Photos and videos














Pinned Tweet

9 Jul 2024
🌟 𝐍𝐞𝐰 𝐚𝐫𝐭𝐢𝐜𝐥𝐞: 𝐭𝐡𝐞 𝐜𝐨𝐧𝐧𝐞𝐜𝐭𝐢𝐨𝐧 𝐛𝐞𝐭𝐰𝐞𝐞𝐧 𝐋𝐨𝐧𝐠 𝐂𝐎𝐕𝐈𝐃, 𝐌𝐲𝐚𝐥𝐠𝐢𝐜 𝐄𝐧𝐜𝐞𝐩𝐡𝐚𝐥𝐨𝐦𝐲𝐞𝐥𝐢𝐭𝐢𝐬 𝐚𝐧𝐝 𝐩𝐨𝐬𝐭-𝐯𝐚𝐜𝐜𝐢𝐧𝐚𝐥 𝐬𝐲𝐧𝐝𝐫𝐨𝐦𝐞𝐬 𝐥𝐢𝐞𝐬 𝐢𝐧 𝐭𝐡𝐞 𝐝𝐞𝐯𝐞𝐥𝐨𝐩𝐦𝐞𝐧𝐭 𝐨𝐟 𝐚𝐧 𝐀𝐮𝐭𝐨𝐢𝐦𝐦𝐮𝐧𝐞 𝐇𝐲𝐩𝐨𝐜𝐨𝐫𝐭𝐢𝐬𝐨𝐥𝐞𝐦𝐢𝐜 𝐒𝐲𝐧𝐝𝐫𝐨𝐦𝐞🌟
🌐 I am excited to share with you our latest paper entitled "𝐇𝐲𝐩𝐨𝐜𝐨𝐫𝐭𝐢𝐬𝐨𝐥𝐞𝐦𝐢𝐜 𝐀𝐒𝐈𝐀: 𝐀 𝐯𝐚𝐜𝐜𝐢𝐧𝐞- 𝐚𝐧𝐝 𝐜𝐡𝐫𝐨𝐧𝐢𝐜 𝐢𝐧𝐟𝐞𝐜𝐭𝐢𝐨𝐧-𝐢𝐧𝐝𝐮𝐜𝐞𝐝 𝐬𝐲𝐧𝐝𝐫𝐨𝐦𝐞 𝐛𝐞𝐡𝐢𝐧𝐝 𝐭𝐡𝐞 𝐨𝐫𝐢𝐠𝐢𝐧 𝐨𝐟 𝐥𝐨𝐧𝐠 𝐂𝐎𝐕𝐈𝐃 𝐚𝐧𝐝 𝐦𝐲𝐚𝐥𝐠𝐢𝐜 𝐞𝐧𝐜𝐞𝐩𝐡𝐚𝐥𝐨𝐦𝐲𝐞𝐥𝐢𝐭𝐢𝐬". In this paper, we explain the links between Long COVID, Chronic Fatigue Syndrome/Myalgic Encephalomyelitis (ME/CFS) and COVID-19 post-vaccine syndromes. 🤔 Stay reading to the end and you will also find our treatment proposal that could improve symptoms.💊
➡️𝐋𝐢𝐧𝐤 𝐨𝐟 𝐨𝐮𝐫 𝐫𝐞𝐯𝐢𝐞𝐰 𝐚𝐫𝐭𝐢𝐜𝐥𝐞: frontiersin.org/journals/imm…
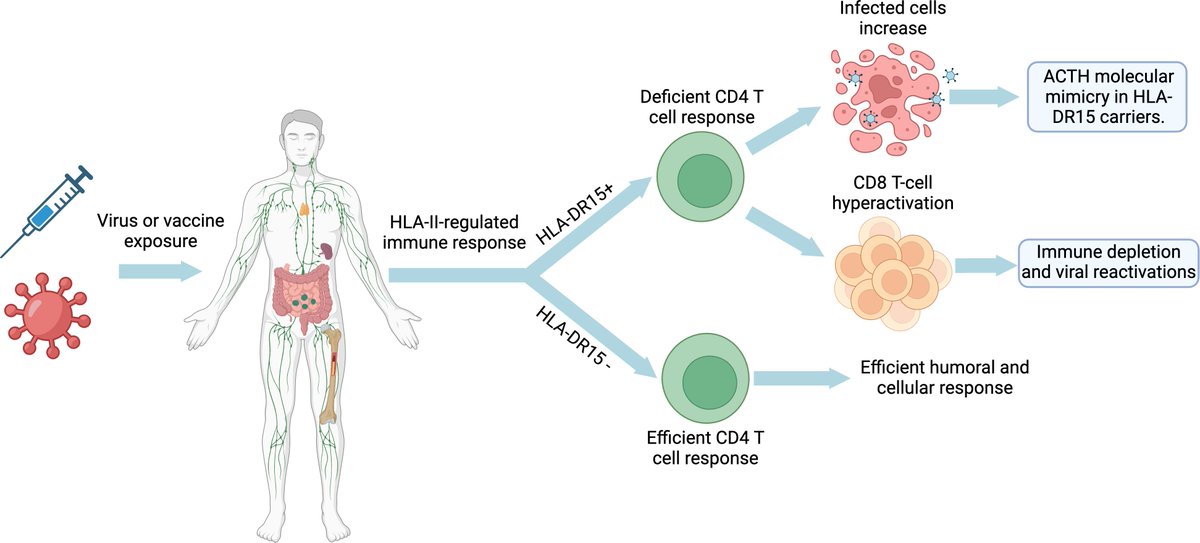
🔍 𝐀𝐛𝐬𝐭𝐫𝐚𝐜𝐭: We present a model for the development of these diseases that involves a complex interplay between immune hyperactivation, autoimmune hypophysitis or pituitary hypofunction, and immune exhaustion. We believe that the starting point is a deficient CD4 T-cell response to viral infections in genetically predisposed individuals (HLA-DRB1). This would follow from an uncontrolled immune response with hyperactivation of CD8 T cells and elevated antibody production, some of which could be directed against self-antigens, triggering autoimmune hypophysitis or direct damage to the pituitary, resulting in decreased production of pituitary hormones, such as ACTH. 🧬
🔬 𝐖𝐡𝐚𝐭'𝐬 𝐭𝐡𝐞 𝐛𝐢𝐠 𝐝𝐞𝐚𝐥?
1️⃣ 𝐑𝐞𝐥𝐚𝐭𝐢𝐨𝐧𝐬𝐡𝐢𝐩 𝐭𝐨 𝐀𝐒𝐈𝐀 𝐒𝐲𝐧𝐝𝐫𝐨𝐦𝐞: We propose that Long COVID, ME/CFS and post-vaccine COVID-19 syndrome could be included in adjuvant-induced autoimmune/inflammatory syndrome (ASIA) due to their similar clinical manifestations and possible relationship to genetic factors, such as polymorphisms in the HLA-DRB1 gene.
2️⃣ 𝐃𝐞𝐯𝐞𝐥𝐨𝐩𝐦𝐞𝐧𝐭𝐚𝐥 𝐌𝐨𝐝𝐞𝐥: We suggest that these diseases begin with a deficient immune response and progress to uncontrolled immune hyperactivation, followed by immune exhaustion, exacerbating symptoms and pathology.
3️⃣ 𝐇𝐲𝐩𝐨𝐜𝐨𝐫𝐭𝐢𝐬𝐨𝐥𝐞𝐦𝐢𝐚: We highlight the decrease in ACTH production and its impact on immune function and clinical symptoms, establishing a direct link with pituitary dysfunction.
4️⃣ 𝐓𝐫𝐞𝐚𝐭𝐦𝐞𝐧𝐭 𝐏𝐫𝐨𝐩𝐨𝐬𝐚𝐥: We propose a treatment approach including antivirals, corticosteroids/ginseng, antioxidants and metabolic precursors to improve symptoms by modulating immune response, pituitary function, inflammation and oxidative stress.
💡 𝐈𝐦𝐩𝐥𝐢𝐜𝐚𝐭𝐢𝐨𝐧𝐬 𝐚𝐧𝐝 𝐂𝐨𝐧𝐜𝐥𝐮𝐬𝐢𝐨𝐧𝐬:
🔹These disorders could have an autoimmune origin against the adenohypophysis.
🔹Treatment with antivirals and corticosteroid replacement therapy in patients with permanent pituitary damage could improve symptoms by addressing immune and hormonal dysfunction.
🧠 𝐓𝐡𝐞 𝐤𝐞𝐲 𝐢𝐬 𝐩𝐢𝐭𝐮𝐢𝐭𝐚𝐫𝐲 𝐝𝐚𝐦𝐚𝐠𝐞: 𝐡𝐨𝐰 𝐝𝐨𝐞𝐬 𝐭𝐡𝐢𝐬 𝐫𝐞𝐥𝐚𝐭𝐞 𝐭𝐨 𝐭𝐡𝐞 𝐝𝐞𝐯𝐞𝐥𝐨𝐩𝐦𝐞𝐧𝐭 𝐨𝐟 𝐌𝐄/𝐂𝐅𝐒, 𝐋𝐨𝐧𝐠 𝐂𝐎𝐕𝐈𝐃 𝐚𝐧𝐝 𝐨𝐭𝐡𝐞𝐫 𝐩𝐨𝐬𝐭-𝐯𝐢𝐫𝐚𝐥 𝐚𝐧𝐝 𝐩𝐨𝐬𝐭-𝐯𝐚𝐜𝐜𝐢𝐧𝐞 𝐬𝐲𝐧𝐝𝐫𝐨𝐦𝐞𝐬?
💉 Certain viruses (and other pathogens) and vaccines can affect the pituitary gland, interfering with cortisol production and triggering a cascade of complex symptoms. In patients with weak HLA-DRB1 alleles, such as DR15, immune hyperactivation can trigger an autoimmune response against ACTH, crucial for cortisol production. This is exactly analogous to how other autoimmune diseases such as multiple sclerosis or lupus develop, where the immune system attacks other antigens in the body, but in the syndromes we are discussing, the autoimmunity is specifically directed against pituitary ACTH.
🦠 This link explains why patients with chronic infections often experience persistent hypocortisolemia, as the pathogen continues to produce ACTH-mimicking antigens, maintaining the active autoimmune response or generates direct pituitary damage. In contrast, patients without chronic infections and with the same weak alleles treated with immune checkpoint inhibitors (ICIs) may develop temporary hypophysitis and similar cortisol deficits, but discontinuation of treatment usually allows recovery.
This also explains why patients experience chronic fatigue, dysautonomia, orthostatic intolerance, exercise intolerance, intolerance to stressful events and mild hypoglycemia due to low cortisol. Cortisol is crucial in providing the body with needed energy and regulating the stress response. When cortisol levels are low, as they are in these syndromes, the body cannot respond effectively to physical and emotional demands.
🩸Cortisol plays a crucial role in maintaining stable blood sugar levels by promoting gluconeogenesis (glucose production) and stimulating the release of stored glucose in the form of glycogen in the liver. When there is insufficient cortisol, the body faces difficulties in increasing glucose levels in demand situations, such as during physical exercise or in response to stress, which can lead to episodes of mild hypoglycemia. In times of fright or anger, adrenaline release may temporarily improve symptoms by temporarily increasing glucose availability, briefly compensating for cortisol deficiency. However, this response does not adequately replace the long-term regulatory functions of cortisol, so symptoms may return once adrenaline subsides.
🏋️♂️ In the case of physical exercise, which naturally increases cortisol levels to mobilize energy and respond to physical exertion, the lack of this hormone limits the body's ability to maintain sustained physical activity. Patients may experience rapid muscle fatigue, feelings of weakness and slower recovery after exercise.
😰 As for stressful events (exams, travel, surgical operations, etc) , cortisol also plays a crucial role in the body's response to emotional or physical stress. When cortisol levels are insufficient, the body has difficulty handling stressful situations effectively. This can manifest itself in an exacerbation of existing symptoms, such as intense fatigue, dizziness, difficulty concentrating and a generalized feeling of malaise.
➡️ For years, many of these patients have been misunderstood and mislabeled as having psychosomatic illness. This is because their symptoms tend to worsen during periods of stress, which has led to the suggestion that the origin of their problems lies in psychological factors. However, the reality is that these patients are not experiencing symptoms due to an underlying psychological disorder, but as a direct result of insufficient cortisol. The lack of this vital hormone prevents the body from adapting and responding appropriately to stress, which perpetuates and aggravates their physical symptoms.
🚨 𝐓𝐡𝐞 𝐃𝐞𝐯𝐞𝐥𝐨𝐩𝐦𝐞𝐧𝐭 𝐨𝐟 𝐀𝐮𝐭𝐨𝐢𝐦𝐦𝐮𝐧𝐢𝐭𝐲 𝐭𝐨 𝐀𝐂𝐓𝐇: 𝐀 𝐏𝐫𝐨𝐜𝐞𝐬𝐬 𝐒𝐢𝐦𝐢𝐥𝐚𝐫 𝐭𝐨 𝐎𝐭𝐡𝐞𝐫 𝐀𝐮𝐭𝐨𝐢𝐦𝐦𝐮𝐧𝐞 𝐃𝐢𝐬𝐞𝐚𝐬𝐞𝐬 𝐬𝐮𝐜𝐡 𝐚𝐬 𝐌𝐮𝐥𝐭𝐢𝐩𝐥𝐞 𝐒𝐜𝐥𝐞𝐫𝐨𝐬𝐢𝐬 🚨
This same mechanism occurs in other autoimmune diseases. Some HLA-II alleles, such as the DR15 variant, are associated with an impaired ability to recognize cells infected with certain pathogens, such as Epstein-Barr virus (EBV). In multiple sclerosis this poor recognition ability specifically affects CD4 T cells, which are crucial for coordinating the immune response. When CD4 T cells cannot correctly recognize infected cells, this leads to hyperactivation of CD8 T cells and an increase in antibodies against the pathogen to compensate for the deficient CD4 T cell response. Without the coordinated help of CD4 T cells, CD8 T cells cannot eliminate all EBV-infected cells, thus never effectively eliminating or controlling the infection and resulting in chronic infection. This results in an increase of infected cells, an exhaustion of CD8 T cells and an increased risk of developing autoimmune diseases, since CD4 T cells, by misrecognizing these viral antigens presented on the HLA-II antigen-presenting cells, can confuse them with the body's own proteins, generating an autoimmune disease. In multiple sclerosis, autoimmunity develops when the EBNA-1 antigen of the Epstein-Barr virus is mistaken for myelin, due to a similar amino acid sequence and molecular mimicry in patients with DR15 alleles. The same could occur in patients with Long COVID, myalgic encephalomyelitis and post-vaccinal syndromes, where autoimmunity against ACTH develops.
#LongCovid #MECFS #Vaccines
42
293
779
124,680
14h
🔴I agree with the general idea that elevated IgG titers to latent or persistent pathogens should not simply be dismissed as meaningless.
But I would add a few mechanistic clarifications.
First, not all herpesviruses establish latency in B lymphocytes as their main reservoir.
EBV is the classic herpesvirus with a major B-cell reservoir. But CMV, HHV-6, VZV and other herpesviruses have different latency biology and different cellular or tissue reservoirs. So it is important not to generalize EBV biology to all herpesviruses.
Second, latency is not simply a state in which infected cells become completely invisible or biologically inactive.
This is especially clear with EBV.
EBV has different latency programs. Latency 0 is the most immunologically silent state, but latency I, II and III still express viral antigens to different degrees, and these infected cells can be controlled by EBV-specific CD4 and CD8 T cells.
For example, latency I, classically seen in Burkitt lymphoma, expresses EBNA1, and EBNA1-specific CD4 T-cell responses are part of the immune control of EBV-infected cells.
But the same general principle applies more broadly: many latent or persistent pathogens are not simply “doing nothing.”
Latently infected or persistently infected cells can express viral non-coding RNAs, microRNAs, latency-associated transcripts or other regulatory RNA species that can modulate immune sensing, inflammation, cell survival, antiviral responses and immune evasion.
In herpesviruses, viral microRNAs and non-coding RNAs can help maintain latency, suppress lytic genes, reduce immune recognition, alter host-cell survival and shape the local inflammatory environment.
Even latently infected EBV cells can express non-coding viral RNAs such as EBERs and EBV microRNAs. These can modulate inflammation, interfere with antiviral sensing, shape B-cell survival and contribute to immune evasion.
EBV also has immunomodulatory strategies, including viral IL-10-like activity mainly associated with lytic or reactivation contexts, which can help dampen antiviral immune responses and reduce immune recognition.
So “latent” does not necessarily mean silent, harmless or irrelevant.
It often means that the virus is being actively contained by immune surveillance.
In healthy people, the key point is not that latent viruses become totally invisible. The key point is that CD4 and CD8 T-cell surveillance, NK-cell function and other antiviral mechanisms keep these reservoirs highly contained.
This is why most healthy carriers do not have constant clinically relevant reactivation. The pathogen is usually controlled, restricted to limited reservoirs, and only reactivates more meaningfully during periods of immune stress, acute infection, immunosuppression or loss of immune control.
Where things become more interesting is in genetically susceptible individuals.
Immunogenetic and evolutionary studies suggest that some ancestral HLA-II haplotypes, especially DR2-DQ6, DR3-DQ2 and DR4-DQ8, have broader or more promiscuous peptide-binding repertoires than many other HLA-II backgrounds.
This broader antigen-presentation capacity may have been advantageous during acute infections, because it allowed stronger and more diverse CD4 T-cell responses against pathogens.
But if the pathogen persists, reactivates, or uses latency/evasion mechanisms, the immune system can remain chronically stimulated.
That chronic stimulation can induce sustained IFN-γ, tissue inflammation, ectopic HLA-II expression in tissues that do not normally present antigen at high levels, and continuous presentation of both pathogen-derived and self-derived peptides.
In that environment, the probability of activating autoreactive T and B cells increases.
This is where HLA background matters.
1/3
Continued in the next post.👇🏻

B cells, go dormant and become undetectable to the immune system. The virus isn't eliminated, just silenced. Then, when we receive a challenge to our immune health (physiological/emotional/physical stress, malnutrition, aging) or we are infected by a pathogen that is known to
11/
3
14
63
4,511
14h
2/3
Molecular mimicry does not happen equally in everyone. It requires that a pathogen-derived epitope and a self-epitope are sufficiently similar, and that the person’s HLA molecules can actually present those epitopes in a way that activates autoreactive clones.
This is one reason why EBV-related molecular mimicry in diseases like multiple sclerosis is strongly shaped by HLA background.
But molecular mimicry is only one mechanism.
Bystander activation is different. That is not simply “the immune system attacking tissue because it is angry.” It is more about chronic inflammation, cytokines, tissue injury, costimulation, loss of local regulation and increased antigen presentation creating an environment where autoreactive lymphocytes that were previously silent can become activated.
Epitope spreading is also related but distinct.
Once inflammation becomes chronic, the immune response can expand from the original antigenic target to additional epitopes from the same antigen, other pathogen antigens, or self-antigens released or modified during tissue damage. Post-translational modifications and neoantigens can also expand the antigenic landscape.
So the process is not just:
infection → molecular mimicry → autoimmunity.
It may be more like:
persistent pathogen/reactivation → chronic antigen presentation → IFN-driven tissue inflammation → ectopic HLA-II expression → molecular mimicry, bystander activation and epitope spreading → autoreactive T/B-cell activation → autoimmunity.
Then a second loop can appear.
Both chronic infection and autoimmunity can drive continuous T-cell activation. Over time, that can lead to exhausted or dysfunctional CD4 and CD8 T-cell phenotypes, including increased PD-1 expression.
Once cellular immune control weakens, latent or persistent pathogens such as EBV, CMV, HHV-6, VZV, parvovirus B19 or other intracellular pathogens may become less controlled.
That can increase the frequency of reactivation, tissue-level infection, infected-cell burden or antigenic exposure.
And when that happens, IgG titers may rise not because IgG is random or irrelevant, but because the immune system is repeatedly seeing antigen again and again.
In reactivation, IgM is often absent or transient because the person already has immune memory. A memory IgG response can rise without a strong new IgM response. So negative IgM does not necessarily rule out reactivation, especially if the process is tissue-localized, intermittent or already being partially contained.
This is why high IgG titers should not be interpreted simplistically.
They do not automatically prove active disease.
But in a chronically ill patient, especially with compatible symptoms, immune dysfunction, HLA susceptibility, abnormal T-cell/NK-cell control, or autoimmune features, they should be the beginning of a deeper investigation, not the end of the conversation.
I would also slightly reframe the “chicken or egg” issue in autoimmunity.
The initial trigger is usually some form of antigenic or inflammatory stimulus: infection, vaccine antigen, allergen, toxin, tissue injury, microbiome antigen, or another immune stressor.
Once autoimmunity develops, reactivations may become both a consequence and an amplifier.
They can result from impaired cellular immune control, but each reactivation can also increase immune activation, expand antiviral T and B cells, increase cytokine release, and indirectly amplify autoreactive clones.
That is why autoimmune flares can coincide with viral reactivation signals.
It is not necessarily because the virus is the only cause at that point.
It may be because reactivation and autoimmunity are now feeding each other.
The same logic may apply to mast-cell activation.
Continued in the next post.👇🏻
1
1
19
490
14h
3/3
Chronic infection, persistent antigen exposure and tissue inflammation can activate mast cells as part of the attempt to recruit and coordinate leukocytes. If the antigenic stimulus is not resolved, mast-cell activation can become chronic and contribute to vascular, neurological, allergic-like and inflammatory symptoms.
So yes, elevated herpesvirus IgG titers in Long COVID, ME/CFS, chronic Lyme/tick-borne illness and other complex chronic illnesses should not be dismissed.
But the interpretation should be more precise:
Not all herpesviruses have the same latency biology.
EBV is the classic B-cell latent herpesvirus, but other herpesviruses have different reservoirs and control mechanisms.
Latency is not always immunologically invisible.
Latent does not mean biologically inactive.
Latent or persistent pathogens can still shape inflammation through non-coding RNAs, microRNAs, latency-associated transcripts and immune-evasion mechanisms.
IgG elevation is not proof by itself of active infection, but it may reflect repeated antigenic exposure or reactivation in the right context.
IgM negativity does not exclude reactivation.
And HLA-DR/HLA-DQ background may be central in determining who converts chronic antigenic stimulation into autoimmunity.
For me, the key model is a vicious circle:
genetic susceptibility persistent antigenic stimulation chronic inflammation T-cell exhaustion impaired viral control reactivation autoimmunity.
That loop may explain why some patients show high IgG titers to latent or intracellular pathogens, not as an isolated laboratory curiosity, but as part of a broader immune-control failure pattern.
2
1
17
342
Jun 8
‼️⚠️I sincerely appreciate that you acknowledged this mistake.
Recognizing errors publicly is not easy, and I respect that.
But I also think this is why I have to ask you to look again at the much bigger issue: the WIRED article itself.
Because the problem is not only one misread study. The broader framing of that article is already causing real harm.
I say this sincerely: I know there is a person behind every account, with their own intentions, limits, mistakes and blind spots. We can all get things wrong. I do not believe the right response is to destroy someone when they are willing to correct mistakes.
But the article needs correction.
Over the last few days, I have received comments and private messages from patients saying that family members, people around them, and even clinicians who do not understand Long COVID are now using this narrative against them.
They are being told that they are not recovering because they do not want to.
Because they are not exercising.
Because they are not doing psychological therapy.
Because they are “stuck” in the wrong mindset.
You may not fully realize the damage that kind of framing can do if you have not lived this disease closely.
For many patients, their environment had finally started to believe them because biomedical research was moving forward. Years of studies showing immune, vascular, autonomic, metabolic and muscular abnormalities were slowly helping people understand that this is a real organic disease.
And then a simple, attractive narrative appears again:
maybe it is mind-body.
maybe patients are afraid of exercise.
maybe recovery is being blocked by beliefs.
That kind of framing can erase years of progress in one family, one workplace, one clinic.
Because when a disease is complex and poorly understood, the easiest story is always the old one: maybe the problem is psychological.
ME/CFS patients have lived with this harm for decades. Many Long COVID patients are now experiencing the same thing.
And this pressure is not harmless.
Patients lose their health.
They lose their jobs.
They lose their social lives.
They lose their independence.
And then, when public narratives suggest that maybe they are not recovering because of their mindset, they can also lose the last thing they had left: being believed and supported.
That pressure can become unbearable.
Some patients end up taking their own lives because they feel abandoned, disbelieved and blamed for an illness they did not choose.
Those lives matter.
They matter as much as yours or mine.
And these patients deserve exactly the same dignity, seriousness and protection that we give to patients with any other recognized disease.
Today, nobody would tell a patient with multiple sclerosis that they remain ill because they do not want to recover, because they do not exercise enough, or because they have not done the right psychological therapy.
Nobody would frame MS as a failure of mindset just because fatigue, stress sensitivity, cognitive symptoms or depression can appear in the disease.
So why is this acceptable with Long COVID or ME/CFS?
This is not about rejecting psychological support.
It is not about denying that the nervous system is involved.
It is not about saying every recovery story is false.
It is about not confusing support with cure.
Not confusing subjective improvement with disease modification.
Not confusing heterogeneous biology with “it might be in your head.”
Not using recovery anecdotes to reframe a post-infectious disease in a way that patients will pay for socially, medically and personally.
I genuinely believe people can reconsider things. None of us has to know everything about every field. Mistakes happen.
But when a mistake has consequences for a vulnerable patient community, correction matters.
The same criticism I have made these days, I would gladly replace with support if you helped correct the framing and the harm caused by the article.
Patients deserve mechanisms.

Would like to note that I made a thread QTing a study that someone said showed 91% of people recovered from PAIS. The study did NOT say that, and I tweeted a misinformed thread that leaned into that incorrect reading. I deleted the tweets, but I will leave this tweet up with the thread I posted so people can see the mistake I made, as well as the original tweet that I QTed.
10
97
359
13,880
Jun 6
⚠️After seeing the response from patients, clinicians and scientists, I think this needs to be said clearly:
Re-promoting an article that has caused so much harm to the Long COVID community is not brave journalism.
It is irresponsible.
This article did not simply “open a difficult conversation.” It amplified a deeply damaging frame: that maybe patients are rejecting the uncomfortable truth that CBT, exercise and “mind-body” approaches are the way forward.
But when you present a complex post-infectious disease through that lens, without properly separating psychological support from biological treatment, you are not helping patients.
You are making their lives harder.
These narratives do not stay inside an article.
They reach families.
They reach employers.
They reach disability assessors.
They reach doctors who already know very little about Long COVID.
They reach people who are looking for an excuse to say: “maybe it is just anxiety,” “maybe you are afraid of exercise,” “maybe you are keeping yourself sick.”
That has consequences.
Social consequences.
Medical consequences.
Workplace consequences.
Psychological consequences.
Patients with Long COVID are already fighting disbelief, lack of biomarkers in routine care, lack of approved treatments, disability, isolation and medical neglect.
Using their suffering to generate clicks while repackaging old psychosomatic narratives in modern “mind-body” language is not courageous.
It is cruel.
The problem is not talking about the nervous system.
The problem is turning nervous system involvement into a story about beliefs, fear, trauma or patients refusing to accept recovery.
The problem is using recovery anecdotes as if they establish causality.
The problem is ignoring those who worsened with exercise.
The problem is presenting PEM caution as dogma.
The problem is being much more generous with “brain retraining” narratives than with the biomedical evidence already showing immune, vascular, autonomic, metabolic and muscular abnormalities in Long COVID.
If a journalist wants to write about Long COVID, they have a responsibility to understand the history of harm done to post-infectious patients.
Because this is not new.
ME/CFS patients have lived this for decades.
“Unexplained” became “psychological.”
“Normal routine tests” became “nothing is wrong.”
“Supportive care” became “cure.”
“Exercise” became “rehabilitation,” even when patients were crashing.
And now the same mistake is being repeated with Long COVID.
Patients are not angry because they reject science.
They are angry because they recognize the pattern.
They have seen what happens when medicine turns biology it cannot yet measure into psychology.
So no, this is not “a way forward.”
A way forward would be stratification, biomarkers, mechanistic trials, antivirals, immunology, dysautonomia research, PEM-safe protocols, autoantibody studies, vascular biology, tissue persistence, metabolism and serious clinical phenotyping.
A way forward would be listening to all patients, including those harmed by exercise and psychologizing narratives.
A way forward would be scientific humility: admitting what we still do not know without turning unexplained biology into psychology.
Not click-driven repetition of the same ideas that have already harmed post-infectious patients for decades.

There might finally be a way forward for long Covid treatment—if only you were allowed to talk about it. wired.com/story/the-painful-…
24
181
653
17,316
Manuel Ruiz retweeted
Jun 1
💊‼️Long COVID could be, at least in some patients, an autoimmune disease occurring alongside chronic infection, antigenic persistence, or viral reactivations.
Okay.
So now what?
Are there treatments?
Can autoantibodies be removed?
Can this autoimmunity be “switched off”?
Are we close to a cure?
The honest answer is this: we are not facing an immediate cure , although possibly a future one, but we are facing a huge shift in how we think about the disease.
Because if part of Long COVID, and possibly also ME/CFS, has an autoimmune basis, then it would no longer make sense to treat it as a diffuse syndrome with no therapeutic direction. We would need to do the same thing we do in other autoimmune diseases: classify patients properly, identify biomarkers, and design a stepwise treatment approach.
Not all patients would have the same mechanism.
Not all patients would respond in the same way.
And not all patients would need the same level of treatment.
But for the first time, a more logical therapeutic map is starting to emerge.
A clearer logic is beginning to appear: identify which patients have a real autoimmune component and, from there, think about which therapies could make sense.
At present, what is accessible would not be a cure, but treatments already known from other autoimmune diseases that could help reduce autoantibody activity, modulate the immune response, or decrease part of the immunological damage.
In the best-case scenario, these would be treatments to improve, stabilize, or reduce autoimmunity. Not to completely “erase” the disease.
If I had to rank the options in an orientative way, thinking about potential usefulness and safety profile, I would do it like this:
1. IVIG
The most reasonable short-term option would probably be IVIG, meaning intravenous immunoglobulin.
IVIG does not directly eliminate all autoantibodies as such, but it can modulate the immune system, compete with pathogenic autoantibodies, block part of the inflammation, and dampen the immunological attack without requiring such intense immunosuppression.
In many autoimmune diseases, it is used precisely because of this immunomodulatory effect. Among this type of therapy, it is probably one of the options with the best balance between potential usefulness and safety profile.
That said, it is not free of side effects, it is expensive, and access is limited.
2. FcRn inhibitors
Next, I would place FcRn inhibitors.
These drugs are very interesting because they reduce the total amount of circulating IgG and, with it, they can also reduce the burden of pathogenic autoantibodies.
The advantage is that they are quite targeted toward the humoral component of autoimmunity, meaning the antibody-mediated part.
The disadvantage is that they are not yet truly established for Long COVID or ME/CFS, and it would still be necessary to demonstrate very clearly which subgroup of patients would benefit the most.
Even so, conceptually, if we are talking about an autoantibody-mediated disease, this is one of the most promising strategies.
3. Immunoadsorption
At the next level, I would place immunoadsorption.
This technique consists of filtering the blood to remove immunoglobulins, especially those that may be participating in the autoimmune process.
It is a more targeted strategy than systemic immunosuppression and, in diseases mediated by autoantibodies, it can make a lot of sense.
Its main limitation is logistical: it is not a simple therapy, it requires specialized centers, and its effects may not last if the immune system continues producing the same autoantibodies afterwards.
(1/3)
🔵Continued in the next post.👇🏻
12
79
257
8,957
Manuel Ruiz retweeted
May 31
⚠️‼️ I came across this news in Spain, and this discourse keeps doing enormous harm.
The post literally says:
“A study from Hospital del Mar reveals a worrying connection between childhood trauma and physical diseases in adult life. From headaches to diabetes, adverse childhood experiences could leave marks on our health. Early prevention has never been so crucial.”
“A study from Hospital del Mar details numerous physical diseases linked to childhood trauma.”
Not because they literally said that most organic diseases are caused by childhood trauma, but because in practice, many of the diseases they attribute to or reduce to trauma end up being organic diseases whose biomarkers have not yet been discovered, or whose proper medical workup was never even done.
And that is the real problem.
For years, many patients are first filtered through the lens of trauma, psychosomatic illness, or somatization, when in reality what they have is an organic disease that has simply not yet been diagnosed. I would even go further and say that this also happens with diseases we now understand well: before reaching the right specialist and before the appropriate tests are performed, many patients are treated as if the origin of their symptoms were psychological or trauma-related, simply because nobody has investigated their case properly.
That is not precision medicine.
That is a way of covering diagnostic ignorance with a psychological narrative.
The history of medicine shows this again and again: many diseases that in their early stages were treated as “nervous,” “hysterical,” or “psychosomatic” later turned out to be well-defined organic diseases. This happened with multiple autoimmune, neurological, and inflammatory diseases. And it is still happening today.
That is why this discourse is so dangerous. Because it not only delays the real diagnosis, but also blames the patient, isolates them, sends them into the wrong clinical pathways, and steals years of serious medical investigation from them.
Most of these patients do not need their illness to be explained through trauma.
They need the right tests, the right specialist, and serious investigation of their real biology instead of convenient theories being projected onto them.
Enough of turning the absence of known biomarkers into a psychological explanation.
What some people today call trauma or somatization too often ends up tomorrow having a name, a mechanism, and a biological marker.
7
32
93
5,559
Jun 3
🔴 I think this is an important point, so I’ll add my perspective here.
I have read the article carefully, and I explain the specific problems I see in this full thread.
My criticism is not that those patients should not be believed.
Of course they should be listened to.
The problem is turning individual recovery stories into a broader framing that risks confusing psychological support with causal treatment, and nervous system modulation with a cure for Long COVID.
Listening to patients who improved does not mean ignoring patients who worsened with exercise, PEM, dysautonomia, autoantibodies, viral reactivations, or objective abnormalities after exertion.
Both groups matter.
But testimonials do not establish causality, and they should not be used to reframe a heterogeneous post-infectious disease as something primarily driven by beliefs, fear, trauma, or “mind-body” loops.
In this thread, I go point by point through what I think the article gets wrong.
If after reading it you still disagree, I would genuinely be interested in hearing your arguments.
👉🏼 x.com/manruipa/status/206189…
Can you point me to specific parts of the article that you think are inaccurate? The central point of this piece is to highlight the voices of extremely sick patients who not only feel dismissed by the medical establishment, but also, when they recovered, were gaslit by people who denied that the programs could have worked for them. Are you saying you don't believe those patients? They are the point of the article.
15
91
2,639
Jun 3
⚠️ I think this is exactly the problem.
When patients with ME/CFS, fibromyalgia or Long COVID defend themselves, it is often framed as “protecting an illness narrative.”
But if your disease were constantly minimized, psychologized, ignored, and left without biological treatments, while patients are told they are exaggerating or inventing symptoms, this would not feel like protecting a narrative.
It would feel like defending the basic right to be taken seriously and treated with the same dignity as patients with any other disease.
These narratives do not only affect medical care. They affect families, friendships, work, disability support, and social isolation.
Patients are not protecting illness.
They are protecting themselves from being abandoned by a system that too often turns unexplained biology into psychology.
I invite you to read the full thread, because I do not think this is a fair defense of the article:
x.com/manruipa/status/206189…

One thing I have noticed about patients with diagnoses such as fibromyalgia, ME/CFS, and increasingly Long COVID is how often the conversation revolves around explaining why they cannot move forward.
There’s an extraordinary effort devoted to protecting the illness narrative.
21
116
523
12,639
Jun 2
(6/6)
The real problem with the article is not that it asks for more research. That is fine.
The problem is that it presents a false dichotomy: either you accept mind-body therapies as a brave possibility, or you are part of a dogmatic community blocking science.
No.
The real scientific position is much simpler:
Long COVID is a real, biological, heterogeneous disease.
The absence of a single test does not invalidate the illness.
CBT can be support, not a causal cure.
Exercise can help some patients, but harm others, especially those with PEM.
Pacing is not fear: it is prevention of worsening.
Dysautonomia can look like anxiety, but have a physiological basis.
Stress can worsen symptoms without being the primary cause.
Neuroplasticity must not be used as a commercial wildcard.
Recovery testimonials do not replace well-designed trials.
And no treatment should be globally recommended without stratifying subgroups.
Medicine has already made this mistake before.
Patients with multiple sclerosis, lupus, autoimmune diseases, ulcers caused by Helicobacter pylori, endometriosis, or ME/CFS were treated for years as exaggerated, hysterical, anxious, or somatizing until technology and research caught up with what patients had been saying all along.
Let us not repeat the same mistake with Long COVID.
The future is not in denying the nervous system.
It is in understanding it properly.
But understanding the nervous system does not mean selling “mind-body” as a cure.
It means studying immunology, dysautonomia, autoantibodies, persistent viruses, endothelium, metabolism, muscle, the HPA axis, mast cells, microbiota, and neuroinflammation.
And then, if a psychological tool helps a patient live better with an organic disease, it is welcome.
But let us call it by its name: support.
Not cure.
Not a total explanation.
Not a substitute for biomedical research.
Not an excuse to prescribe exercise to someone with PEM.
Not another elegant way of saying “it is all in your head.”
Because it is not all in the head.
It is in the immune system.
It is in the vascular system.
It is in the autonomic nervous system.
It is in metabolism.
It is in muscle.
It is in the tissues.
It is in a biology we are still learning how to measure.
And precisely for that reason, we need more science, not more gurus.
9
40
203
3,622
Jun 2
If this topic matters to you, feel free to visit my profile. I write about Long COVID, ME/CFS, autoimmunity, dysautonomia and the immunology behind complex chronic diseases.
My goal is to make complex biology easier to understand, because patients deserve mechanisms, not stigma.
8
5
132
1,083
Jun 2
(4/6)
So in a chronic post-infectious disease with flares, PEM, inflammation, dysautonomia, and possible immunometabolic dysfunction, exertion can be an additional biological burden. It can increase oxidative stress, energy demand, sympathetic activation, and symptomatic worsening in certain patients.
It is not fear of exercise.
It is physiological intolerance to exertion.
And when thousands of patients say that exercise makes them worse, they are not “reading too much on the internet.” Many have tested it in their own bodies before they even knew what PEM was.
Another very problematic point:
“Talking about psychology can destroy your career.”
No. The problem is not talking about psychology.
The problem is using psychology to cover up medical ignorance.
Patients do not reject psychological support. They reject being told that their symptoms come from childhood trauma, anxiety, depression, false beliefs, or fear of movement when they have postural tachycardia, orthostatic intolerance, PEM, viral reactivations, autoantibodies, immune abnormalities, or muscle damage after exertion.
One thing is psychologically supporting a person with a chronic illness.
Another thing is turning the illness into something psychological.
CBT can help someone cope with a real disease. It can help with insomnia, adaptation, loss of a previous life, grief, secondary anxiety, boundary-setting, pacing, or the understandable fear that comes after worsening many times.
But CBT does not remove autoantibodies.
It does not eliminate viral reservoirs.
It does not clear persistent antigens.
It does not repair endothelial damage.
It does not cure exertion-induced myopathy.
It does not turn autoimmune dysautonomia into health.
It can be support. Not causal cure.
And that nuance is everything.
The same applies to reducing stress, resting, or pausing work.
If a patient improves after resting, taking beta-blockers, reducing workload, doing very gradual walks, and receiving cognitive support, that does not prove Long COVID is psychological. It may prove exactly the opposite: that their autonomic nervous system was overloaded and that by reducing sympathetic load, controlling heart rate, and spacing activities, they can tolerate activity better.
That is biology.
If a patient with dysautonomia improves by reducing stimuli, sleeping better, avoiding heat, controlling heart rate, using beta-blockers, or spacing tasks, it does not mean “the mind cured it.” It means you reduced physiological triggers.
🔵Continued in the next post.👇🏻
1
22
130
2,110
Jun 2
(5/6)
The article constantly confuses “non-pharmacological interventions” with “mind-body therapies.”
Not everything non-pharmacological is psychological.
Rest is not psychotherapy.
Pacing is not positive thinking.
Reducing sympathetic load is not placebo.
Cognitive speech therapy is not “healing with the mind.”
Controlling heart rate in POTS is not pseudoscience.
Avoiding crashes is not fear of movement.
Adapting activity to PEM is not catastrophizing.
It is applied physiology.
And if we want to properly study the role of psychological support, let us do it properly.
For example: select patients with Long COVID and clear dysautonomia. Measure POTS, heart rate variability, orthostatic intolerance, autoantibodies against autonomic receptors, sleep, cortisol, inflammation, and PEM. Give one group tools for activity regulation, breathing, pacing, reduction of sympathetic load, and management of physiological stress. Compare with controls. Measure objective and subjective outcomes.
That would be interesting.
But saying “mind-body” without defining subgroups, without a mechanism, without biomarkers, without concrete goals, and without separating support from causality opens the door to gurus, coaches, and expensive programs for desperate patients.
And that also needs to be discussed.
Because the article criticizes those who sell microclots, antivirals, or supplements without enough evidence, but seems much more lenient toward those who sell neuroplasticity, brain retraining, and mind-body healing.
Pseudoscience does not only wear an alternative medicine coat. Sometimes it also speaks the language of neuroscience.
The term “neuroplasticity” does not make an intervention scientific. Neuroplasticity exists, yes. But using a real word does not validate every commercial program built around it.
Immunology also exists. And that does not mean every “immune-modulating” supplement cures an autoimmune disease.
The question is not whether the brain can influence symptoms. Of course it can.
The question is whether that intervention proves it can modify the disease, in which subgroup, through what mechanism, with what safety, for how long, and with what objective outcomes.
Without that, it is not medicine. It is narrative.
The article ends by arguing that “believing all patients” also means believing those who say they improved with mind-body therapies.
Agreed. We should listen to them.
But listening does not mean concluding causality.
Listening does not mean extrapolating to everyone.
Listening does not mean turning testimonials into treatment.
Listening does not mean ignoring those who worsened.
Listening does not mean recommending exercise to patients with PEM.
Listening does not mean psychologizing post-infectious diseases all over again.
Believing patients also means believing those who say:
“Every time I exercise, I crash for days.”
“My body does not regulate heart rate.”
“Heat destroys me.”
“I wake up with tachycardia.”
“Antihistamines help me.”
“Physical stress triggers flares.”
“After a viral infection, I never returned to normal.”
“They told me it was anxiety, and years later they found autoimmunity.”
Patients are not an obstacle to science. Very often, they are the ones who give the first clues.
If you listen to patients, patterns appear: PEM, POTS, orthostatic intolerance, worsening with heat, food intolerances, responses to antihistamines, viral reactivations, sensitivity to stimuli, crashes after cognitive exertion, flares after infections, improvement with real rest, worsening from overload.
That is not noise. Those are clinical data.
🔵Continued in the next post.👇🏻
2
19
142
1,623
Jun 2
(3/6)The article also presents “astonishing” recoveries as if they were evidence in favor of brain retraining.
But this is scientifically weak too.
In many post-infectious diseases, there is a window in which some patients improve over time. This happens after viral infections such as EBV. Some people have symptoms for months after mononucleosis and then recover. Others do not recover and develop persistent symptoms, autoimmunity, or post-infectious syndromes.
If a person improves at 6, 9, or 12 months while doing a “brain retraining” program, that does not prove the program cured them. It may have coincided with natural recovery, pacing, rest, reduced workload, symptomatic treatment, beta-blockers, antihistamines, aspirin, LDN, antivirals, better sleep, less physical stress, fewer infections, or simply fluctuation of the disease.
Testimonials matter. But they do not replace causality.
And you cannot use patients who improve to invalidate patients who worsen with exercise.
Another point from the article:
“If exercise did indeed trigger post-exertional malaise in most patients, this level of caution would be warranted.”
This approach is extremely dangerous.
First, because post-exertional malaise does not always appear during exercise. Many times it appears 24, 48, or 72 hours later. If you perform a test in a controlled setting and only look at the immediate response, you may not be capturing the main problem.
Second, because Long COVID is heterogeneous. The fact that one subgroup tolerates rehabilitation does not mean that another subgroup with PEM, POTS, or autonomic autoimmunity can tolerate it.
Third, because there are already studies showing objective abnormalities after inducing PEM in Long COVID patients, including muscle abnormalities, metabolic alterations, and worsening muscle damage after exertion.
That is why the cautious recommendation is not “everyone should exercise.”
The cautious recommendation is: first stratify.
Does the patient have PEM?
Do they have POTS?
Do they have dysautonomia?
Do they have autonomic autoantibodies?
Do they have muscle damage or metabolic intolerance to exertion?
Are they in a flare?
Are they mild, moderate, severe, or bedbound?
Do they worsen 24–48 hours after physical or cognitive activity?
Without answering that, recommending exercise globally is not personalized medicine. It is playing roulette with vulnerable patients.
The sentence “not exercising increases the risk of cardiovascular disease, metabolic disease, diabetes, depression, or Alzheimer’s” is true in the general population.
But using it to pressure patients with PEM is a bad extrapolation.
We could also say that sunlight is necessary for vitamin D. But that does not mean you send someone with photosensitive lupus into the sun during a flare.
We could also say that exposure to pollen is harmless for many people. But that does not mean you run a trial putting patients allergic to olive pollen in an olive grove to prove to them that nothing happens.
We could also say that movement is healthy. But if a person with severe Long COVID worsens every time they exceed their limit, the scientific thing to do is to investigate why, not tell them they are deconditioned.
When you have an acute flu, the doctor does not say: “go for a run, otherwise you will become deconditioned.”
They say: rest, hydration, and caution.
🔵Continued in the next post.👇🏻
2
20
137
1,783
Jun 2
‼️⚠️Please read this until the end.
A widely shared article has presented a deeply misleading view of Long COVID, suggesting once again that cognitive behavioral therapy, exercise, and “mind-body” approaches may be the uncomfortable truth patients refuse to accept.
This needs to be challenged.
Not because the nervous system does not matter.
Not because psychological support cannot help.
But because confusing support with cure, physiology with psychology, and heterogeneity with “it might be in your head” is exactly how medicine has harmed post-infectious patients for decades.
There are articles about Long COVID that look like science journalism, but in reality they repackage, in modern language, a very old idea: if we do not fully understand a disease, maybe the problem is in the patient’s mind.
And that is not science. That is repeating history.
The article begins with a striking sentence:
“There isn’t a single approved pharmaceutical treatment, not even a test to verify the presence of the illness.”
This may sound forceful, but it is a very misleading way of presenting the problem.
The fact that there is still no drug specifically approved for Long COVID, or a single diagnostic test, does not mean that “nothing has been found.” It means that we are dealing with a heterogeneous disease, probably with several biological subgroups, and that medicine has not yet converted those findings into validated clinical tools.
“No single diagnostic biomarker” is not the same as “no biology.”
In just a few years, immunological, vascular, neurological, endocrine, and metabolic abnormalities have been described in subgroups of Long COVID patients: autonomic dysfunction, herpesvirus reactivations such as EBV/HHV-6, alterations in the cortisol axis, autoantibodies against GPCR receptors — including adrenergic and muscarinic receptors — persistent viral antigens, endothelial damage, muscle abnormalities after exertion, mitochondrial dysfunction, persistent inflammation, and differential immune changes.
Is everything settled? No.
Does that mean it is psychological? Also no.
Science does not work like that. Multiple sclerosis did not stop existing before we had MRI. Many autoimmune diseases do not show up in routine blood tests. If a complete blood count, a basic biochemistry panel, or an X-ray comes back “normal, normal, normal,” that does not prove the absence of disease. It only proves that you are looking with inadequate tools.
One of the article’s most serious mistakes is this: it confuses the absence of a simple clinical test with the absence of organic disease.
And that mistake has caused harm for decades.
The article also says:
“Almost $2 billion and half a decade of international effort have yielded little more than hypotheses about micro blood clots and spike proteins and mitochondrial dysfunction.”
No. That is not correct.
A hypothesis is a provisional explanation. But when you compare patients and controls and find significant differences in muscle tissue, metabolism, response to exertion, immune biomarkers, viral antigens, autoantibodies, or vascular dysfunction, you are no longer talking about “little more than hypotheses.” You are talking about lines of biomedical evidence that still need to be organized, replicated, stratified, and translated into treatments.
That is not scientific failure. That is research into a complex and new disease.
🔵Continued in the next post.👇🏻
(1/6)
Six years since the height of the pandemic, the scientific community remains baffled by long Covid. But there might finally be a way forward for long Covid treatment—if only you were allowed to talk about it.
wired.com/story/the-painful-…
35
215
686
32,954
Jun 2
(2/6)The real problem is not that “there is nothing.” The problem is that Long COVID is not one single thing.
There are patients with an ME/CFS phenotype and post-exertional malaise.
There are patients with POTS/dysautonomia.
There are patients with lung damage.
There are patients with viral reactivations.
There are patients with immunological abnormalities.
There are patients with autoantibodies.
There are patients with neurocognitive symptoms.
There are patients with intolerances, MCAS-like symptoms, digestive problems, or worsening with heat.
And there are patients with combinations of all of the above.
If you put everyone into the same bag, any study will come out confusing.
But that heterogeneity does not justify returning to the discourse of “cognitive behavioral therapy and exercise.”
In fact, the most dangerous sentence in the article is the underlying idea: because some patients improve with “mind-body” therapies, maybe the Long COVID community is rejecting an uncomfortable truth.
No.
What the community rejects is not the idea that the nervous system participates in the disease. Of course it participates. What is rejected is using that fact to turn an organic disease into a problem of beliefs, trauma, fear of movement, or a “mental loop.”
Because there is a huge difference between these two sentences:
1. “The autonomic nervous system is altered, and we can help the patient modulate symptoms while we investigate and treat the biological cause.”
2. “Your brain is stuck in a fight-or-flight loop, and you need to retrain it in order to recover.”
The first is supportive medicine.
The second can very quickly become pseudoscience.
And this matters.
Many Long COVID patients describe postural tachycardia, heat intolerance, exertion intolerance, insomnia, tremors, shortness of breath, dizziness, food intolerances, worsening after physical or emotional stress, and the feeling of being in “fight or flight.”
But that state does not have to be psychological.
It can be explained biologically.
If a subgroup of patients develops autoantibodies against autonomic receptors — for example muscarinic or adrenergic receptors — you can alter the sympathetic/parasympathetic balance. If you reduce the parasympathetic “brake” or alter vasoregulation, the patient may live with tachycardia, physiological hyperarousal, poor orthostatic tolerance, fatigue, dizziness, stress intolerance, and a constant feeling of activation.
From the outside, that can look like anxiety.
But it is not the same as primary anxiety.
It is altered autonomic physiology.
And this is the major flaw of many “mind-body” approaches: they observe a real nervous system symptom, but they do not understand why it is happening. So they fill the gap with attractive words: neuroplasticity, trauma, survival brain, mind-body loop, trapped nervous system.
But if you do not identify the mechanism, you are only renaming ignorance.
🔵Continued in the next post.👇🏻
4
27
173
6,476
Jun 1
‼️That state can also be explained biologically. If some patients develop autoantibodies against muscarinic receptors such as M3, and possibly other autonomic targets, parasympathetic signaling may be impaired. In simple terms, the “brake” of the autonomic nervous system does not work properly, so the body can remain stuck for longer in a fight-or-flight state. That could help explain dysautonomic flares without needing to reduce everything to a purely psychological or abstract brain-loop model.
Becca Kennedy, the former lead of a multistate long Covid specialty group, has a controversial theory. She believes long Covid belongs to a family of chronic conditions that occur when the brain gets stuck in a feedback loop of fight or flight.
wired.com/story/the-painful-…

5
23
95
4,129
Jun 1
💊‼️Long COVID could be, at least in some patients, an autoimmune disease occurring alongside chronic infection, antigenic persistence, or viral reactivations.
Okay.
So now what?
Are there treatments?
Can autoantibodies be removed?
Can this autoimmunity be “switched off”?
Are we close to a cure?
The honest answer is this: we are not facing an immediate cure , although possibly a future one, but we are facing a huge shift in how we think about the disease.
Because if part of Long COVID, and possibly also ME/CFS, has an autoimmune basis, then it would no longer make sense to treat it as a diffuse syndrome with no therapeutic direction. We would need to do the same thing we do in other autoimmune diseases: classify patients properly, identify biomarkers, and design a stepwise treatment approach.
Not all patients would have the same mechanism.
Not all patients would respond in the same way.
And not all patients would need the same level of treatment.
But for the first time, a more logical therapeutic map is starting to emerge.
A clearer logic is beginning to appear: identify which patients have a real autoimmune component and, from there, think about which therapies could make sense.
At present, what is accessible would not be a cure, but treatments already known from other autoimmune diseases that could help reduce autoantibody activity, modulate the immune response, or decrease part of the immunological damage.
In the best-case scenario, these would be treatments to improve, stabilize, or reduce autoimmunity. Not to completely “erase” the disease.
If I had to rank the options in an orientative way, thinking about potential usefulness and safety profile, I would do it like this:
1. IVIG
The most reasonable short-term option would probably be IVIG, meaning intravenous immunoglobulin.
IVIG does not directly eliminate all autoantibodies as such, but it can modulate the immune system, compete with pathogenic autoantibodies, block part of the inflammation, and dampen the immunological attack without requiring such intense immunosuppression.
In many autoimmune diseases, it is used precisely because of this immunomodulatory effect. Among this type of therapy, it is probably one of the options with the best balance between potential usefulness and safety profile.
That said, it is not free of side effects, it is expensive, and access is limited.
2. FcRn inhibitors
Next, I would place FcRn inhibitors.
These drugs are very interesting because they reduce the total amount of circulating IgG and, with it, they can also reduce the burden of pathogenic autoantibodies.
The advantage is that they are quite targeted toward the humoral component of autoimmunity, meaning the antibody-mediated part.
The disadvantage is that they are not yet truly established for Long COVID or ME/CFS, and it would still be necessary to demonstrate very clearly which subgroup of patients would benefit the most.
Even so, conceptually, if we are talking about an autoantibody-mediated disease, this is one of the most promising strategies.
3. Immunoadsorption
At the next level, I would place immunoadsorption.
This technique consists of filtering the blood to remove immunoglobulins, especially those that may be participating in the autoimmune process.
It is a more targeted strategy than systemic immunosuppression and, in diseases mediated by autoantibodies, it can make a lot of sense.
Its main limitation is logistical: it is not a simple therapy, it requires specialized centers, and its effects may not last if the immune system continues producing the same autoantibodies afterwards.
(1/3)
🔵Continued in the next post.👇🏻
12
79
257
8,957
Jun 1
(3/3)
So, if we summarize it simply, the therapeutic map would look something like this:
First, IVIG.
Then, FcRn inhibitors.
Then, immunoadsorption or plasmapheresis.
Later, anti-CD20 in very selected cases.
And in the long term, deep reset strategies such as CAR-T.
Obviously, this is not a fixed rule and would not apply equally to all patients. It would depend on the immunological subtype, biomarkers, severity, risks, and whether an autoimmune mechanism is truly confirmed in each case.
But as a general map, this would be a reasonable logic if the autoimmune model is confirmed.
Now, there is one idea that, for me, is inseparable from all of this:
If these diseases combine autoimmunity, antigenic persistence, and viral reactivations, then it would make no sense to treat only the autoimmune layer.
Because if you immunomodulate, reduce IgG, deplete B cells, or reset the immune system without controlling the infectious component, you could worsen the very component that keeps feeding the problem.
That is why, in my opinion, any therapy directed at autoimmunity in these patients should be accompanied by a well-designed antiviral strategy before, during, and after treatment.
Before, to reduce viral reactivations or active antigenic burden that may already be present.
During, to prevent immunomodulation, IgG reduction, B-cell depletion, or immune reset from facilitating new reactivations.
And after, to try to prevent the system from becoming reactivated against the same infectious stimulus that may have been maintaining the problem from the beginning.
This would be especially important in therapies such as IVIG, FcRn inhibitors, immunoadsorption, plasmapheresis, anti-CD20 therapies, or future CAR-T-like strategies, because all of them modify, in one way or another, the balance between autoimmunity, antibodies, B cells, and antiviral control.
So the answer to “what do we do now?” would be this:
In the short term, better stratify patients and use existing autoimmune therapies to reduce the disease burden.
In the medium term, incorporate more targeted therapies against pathogenic IgG.
In the long term, aim for a true immunological reset.
And throughout the whole process, never forget the infectious component and viral reactivations.
Because if Long COVID and part of ME/CFS truly are autoimmune/post-infectious diseases, then medicine will also have to stop treating them as if they were one single thing.
It will not be enough to switch off inflammation.
It will not be enough to suppress autoantibodies.
And it will not be enough to give an isolated antiviral.
We will need to treat the autoimmunity, yes.
But also the chronic stimulus that may be maintaining it.
1
5
40
1,041
Jun 1
If you are interested in immunology, autoimmunity, Long COVID, ME/CFS, chronic infections, and post-infectious diseases, you can find more posts like this on my account.
I’m just trying to explain complex biological mechanisms in a simple way, so more people can understand what may be happening behind these diseases.
Because understanding them better is the first step toward taking them seriously.
5
3
34
638