
A new relationship with your health. Agent-native · Science-first
Joined February 2026
- Tweets 81
- Following 42
- Followers 13,493
- Likes 59
21 Photos and videos









Pinned Tweet

Mar 27
Years of meals logs on one app. Sleep tracked on another. Lab results in a PDF somewhere. Apple Watch on your wrist. And you’re still copy-pasting screenshots into ChatGPT at midnight asking “Why do I feel like this?”
That’s the gap we built Turri for.
More than a tracker. More than a chatbot. More than any single health app. 10 AI health agents that work across all your data at once — find the patterns none of your apps ever connect, search the literature for why, and design experiments you can actually run on yourself.
Open source. Runs on your device. Your data never leaves.
🔗compound-longos-clawskill.ve…
26
7
33
25,639
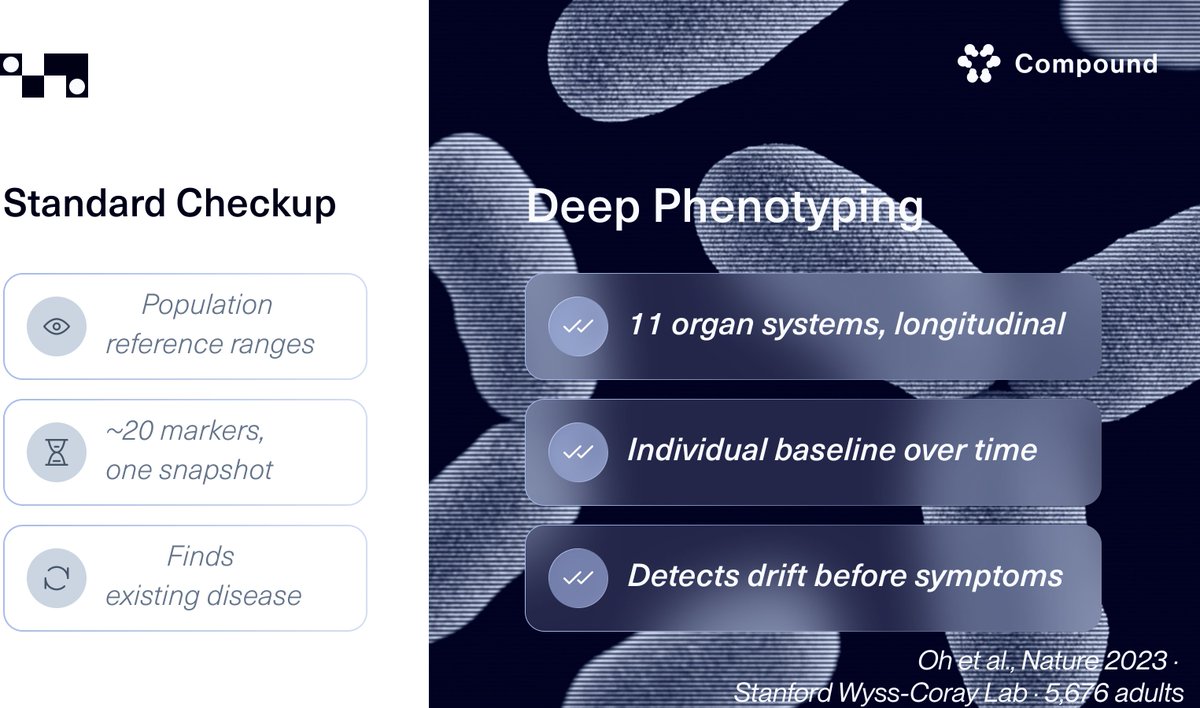
The aging biomarker field has a measurement problem that nobody talks about. Over the past decade, we've accumulated dozens of epigenetic clocks, each trained on different cohorts, different preprocessing pipelines, different endpoints. Horvath, GrimAge, PhenoAge, DunedinPACE. All claim to measure biological age. But because none were ever tested head to head on the same dataset, the honest answer to "which one works best" has been: we don't actually know.
The Biomarkers of Aging Challenge, now published in Nature Aging, took the same approach that transformed protein structure prediction. CASP gave us AlphaFold. This challenge gives aging measurement its first real benchmarking arena. Five hundred individuals, ages 18 to 99, DNA methylation profiles paired with real health outcomes, all run through a single standardized platform. Open to anyone. The winning models identified novel methylation signatures and outperformed existing biomarkers by combining advanced machine learning with biological domain knowledge that no single lab would have deployed alone.
The question that matters most in longevity science right now isn't whether we can measure aging. It's whether we can agree on how to measure it. This is the infrastructure that makes agreement possible. 🧬
pubmed.ncbi.nlm.nih.gov/4227…
16
Jun 11
Do mice and humans actually age by the same dynamics?
A large share of longevity intervention research is built on mouse models, and the working assumption is that the same basic process plays out across species, just at different speeds. A new analysis in Nature Aging from Uri Alon's group at Weizmann suggests the picture is more nuanced. They fit a damage accumulation model across nine species and found two distinct regimes. In mice, damage production overwhelms clearance almost from the start, so damage accelerates like free fall. In humans, production and clearance stay in dynamic balance for decades. The system only tips at very old age.
If the dynamics are that different, how confidently can we extrapolate a mouse longevity result to ourselves? The paper doesn't answer that. But it's a question worth sitting with.
16
Jun 9
Aging research has spent a decade adding to the list. Nine hallmarks became twelve. Twelve became fourteen. A new Cell review makes the opposite move and asks whether they all converge on the same process.
The process is mesenchymal drift. As cells age, they don't just lose function or accumulate damage randomly. They move in a specific direction, progressively acquiring mesenchymal features across tissues and organ systems. Lu, Tu, Ma et al. mapped this signature across over 40 human tissues and 20 diseases. The pattern is consistent: mesenchymal gene programs intensify with age, and the degree of drift predicts disease progression and mortality. This is not a metaphor. It is a measurable trajectory.
The intervention logic shifts accordingly. If the hallmarks are independent, you need 12 drugs. If they converge on one upstream process, you need one good way to measure the drift and one way to reverse it. Partial reprogramming appears to do exactly that, restoring cellular identity before cells have drifted too far. The field went from cataloguing what goes wrong to asking whether there's a single thing going wrong in a single direction. 🧬
1
72
Jun 9
20
Jun 6
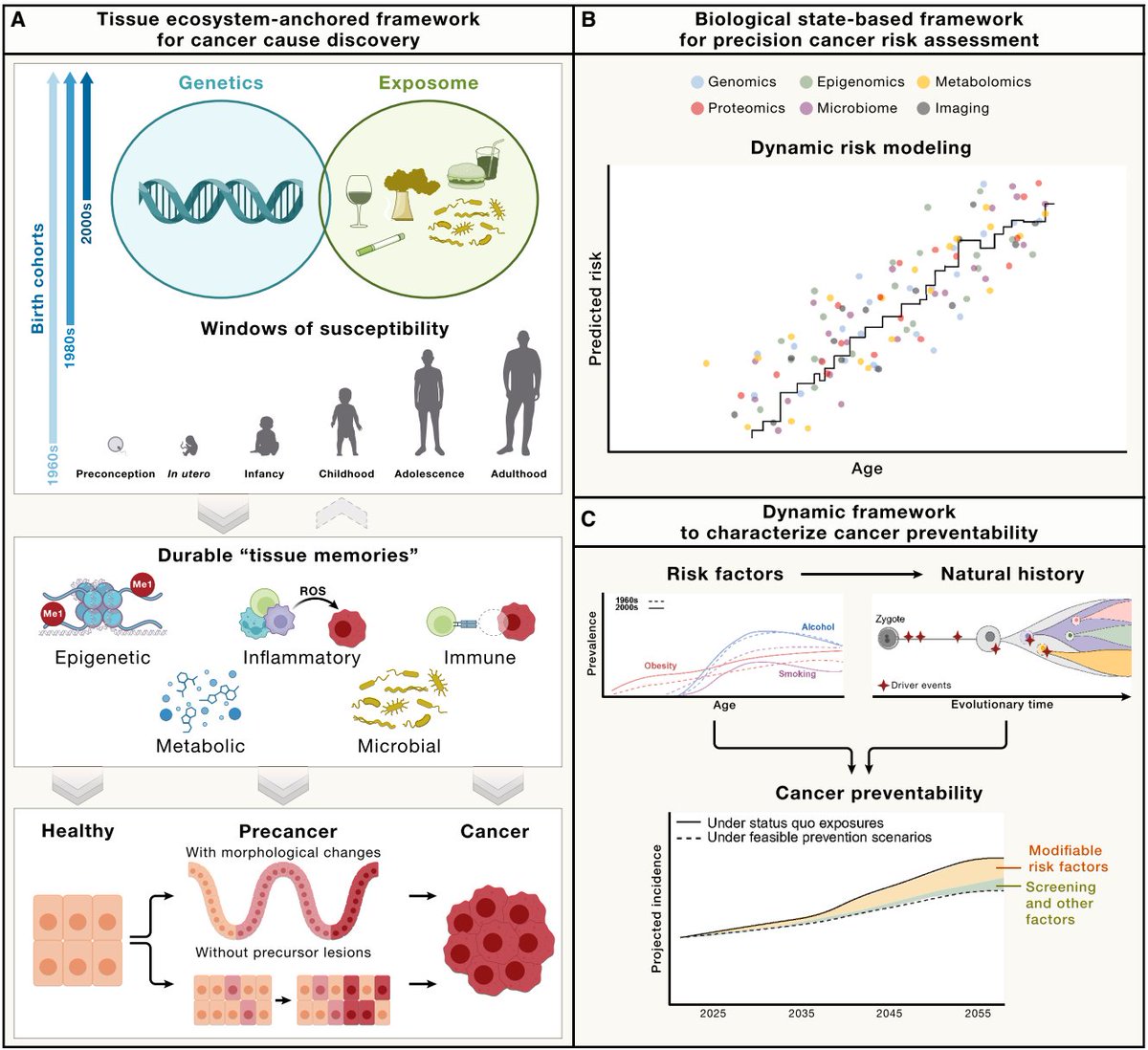
Statins work because we solved blood-based risk stratification for cardiovascular disease decades ago. Measure LDL, treat if it's high, prevent the event. Cancer prevention has never had that kind of molecular handle.
A 14-protein plasma signature, trained on 48,000 UK Biobank participants and validated across eight independent cohorts, now predicts lung cancer more than five years before diagnosis. The proteins are not shed by a tumor. They reflect an inflammatory pre-disease state in lung tissue, upstream of cancer itself.
When Swanton's team re-analyzed the CANTOS trial, identifying high-signature individuals for anti-inflammatory treatment cut the number needed to treat to 55. That is statin-level precision applied to cancer prevention.
The pattern keeps repeating across disease categories: blood-based molecular surveillance, applied early enough, turns detection into prevention. The measurement science is arriving. The system to deliver it to individuals is not.
Pandya, Zagorulya et al. Cell, 2026. 🧬
1
38
Jun 3
Does intermittent fasting work differently for men and women?
Yes. A new study tracking 528 mice across their entire lifespan found that 8-hour time-restricted feeding extended male median lifespan by 12%, but had no significant effect on female lifespan. Both sexes gained healthspan benefits, with females actually showing more durable improvements in frailty, body composition, and behavioral rhythms relative to their total lifespan. Same window, same diet, different biological outcomes.
One important caveat the authors flag: males in the 8-hour group spontaneously ate less than controls. So the lifespan signal may be partly driven by mild caloric restriction, not time restriction alone. The study design provided enough food for ad libitum intake within the window, but the compressed schedule meant males simply didn't finish. Females did not show the same spontaneous reduction.
What this actually tells you is that the question "does fasting work" is underspecified. Work for what? Healthspan or lifespan? And for whom? The answer splits by sex, by endpoint, and possibly by whether the fasting window incidentally restricts calories. One protocol, at least three different biological stories.
2
1
1
75
Jun 3
25
May 29
Gladyshev Lab just published the largest transcriptomic aging study to date in Nature. 11,000 gene expression profiles across four species (mouse, rat, macaque, human), 25 tissue types, 79 interventions.
What they built: a set of multi-species, multi-tissue clocks that predict not just chronological age, but expected mortality. The mortality clock outperformed dedicated lifespan clocks at distinguishing animals on longevity interventions from controls. Aging signatures were conserved across species and across 48 of 49 cell types tested. Two genes, CDKN1A and LGALS3, were validated against mortality and multimorbidity in the UK Biobank.
What changes the picture: aging is modular. Gene expression clusters into 23 co-regulated modules, each with its own clock, each responding to different interventions. Caloric restriction slows metabolic modules. Chronic disease accelerates inflammatory ones. Klotho deficiency hits mitochondria. One biological age number was always a compression of 23 different trajectories.
nature.com/articles/s41586-0…
1
1
38
May 27
Aging just quietly became something pharma companies bid on. This week Daewoong, a Korean drugmaker, won an auction for Turn Bio's cellular reprogramming platform, built to reset aging inside cells.
That kind of therapy is a decade out and aimed at specific diseases. What's available right now is more basic and almost entirely ignored. Most people still can't see where their own aging is heading. Waiting on a future cure while flying blind on your current trajectory is the gap worth closing first.
1
61
May 27
Source on the Daewoong–Turn Bio deal, for anyone who wants the full picture: koreaherald.com/article/1074…
49
May 25
Most people treat sleep as a habit to discipline. You chase more hours and a better score in the morning, and you grade yourself on whether you hit them. The frontier of sleep science has quietly moved somewhere else. Sleep is not a behavior to fix. It is one of the highest-resolution biological signals your body produces, every night, for free, and most people read it at the shallowest possible layer.
Here is what the research actually says.
Start with the one metric everyone optimizes, the number of hours. In UK Biobank data, that is not even the number that best predicts mortality. The regularity of your sleep timing predicts how long you live better than its duration does (Windred et al., 2024). The thing most people track turns out not to be the thing that matters most.
The reason is that sleep carries far more information than a single number. The Human Phenotype Project recorded 448 distinct sleep parameters from more than 16,000 nights of home monitoring across 6,366 adults, and from them predicted body measures spanning 15 of the 16 organ systems they examined, with visceral fat the single strongest correlate. At the data level, a night of sleep is a readout of how your whole body is doing.
There is real biology underneath this. During deep sleep the brain runs a clearance system that flushes metabolic waste, including the proteins implicated in neurodegeneration. Work in Cell mapped how that clearance is driven, though the clearest mechanistic evidence is still in animal models (Hauglund et al., 2025). Deep sleep is doing measurable biological work even while everything looks switched off.
The relationship also has an optimum. In large UK Biobank analyses, cognitive and mental health outcomes peak around seven hours, with both short and long sleep doing worse (Li et al., 2022). A signal with an optimum is exactly the kind of thing a single dashboard number fails to capture.
And the baseline you measure against is not universal. Across 220,000 wearable users in 35 countries, East Asians slept shorter, later, and with lower efficiency than people in Western Europe and North America (Willoughby et al., 2023). The familiar advice to get eight regular hours was calibrated to a different population than the one a lot of people in this region actually live in.
Put it together. The hours you track are not the metric that matters most. The signal is dense with information, it does measurable biological work, it has a sweet spot rather than a maximum, and its baseline shifts with where you live. So the useful question stops being whether you slept enough last night. It becomes what your sleep is telling you about where your metabolism and your heart are heading, and whether anything is reading it continuously. The shift from a score to a signal is the whole game. 🧬
1
62
May 22
This is the line worth sitting with: metabolic, cardiovascular, and mental health were never separate systems. We just measured them separately.
A single intervention moving all three isn't really a surprise. It's a hint that the body keeps one set of books, and most health tools are still reading one page at a time.
May 19

We may be underestimating how deeply connected human biology really is.
A large new study published in @Lancet Psychiatry found associations between GLP-1 use and lower risks of depression, anxiety, substance use disorders, and other psychiatric outcomes across nearly 95,000 people.
We are still early in understanding what is driving this. Some of it is likely indirect: better metabolic health, weight loss, lower inflammation, improved sleep, mobility, and the psychological impact of finally feeling healthier again.
But there is also growing evidence that GLP-1s interact more directly with the brain’s reward and impulse pathways in ways that extend beyond appetite regulation.
For decades, medicine has treated metabolic health, cardiovascular health, and mental health as separate systems. Biology may not work that way.
The same pathways tied to obesity and insulin resistance may also influence addiction, compulsive behavior, mood, and cognitive health.
There is still a lot we need to learn, but the potential impact of these medicines keeps getting bigger - especially as they grow increasingly more affordable.
#Healthstack #Healthspan #Longevity #PatientBack
inc.com/bill-murphy-jr/a-mas…
2
59
May 21
Metabolic aging gets blamed on the mitochondria.
A new paper in Nature Aging puts the first failure one step upstream, in an organelle most people never think about.
Peroxisomes break down hard-to-burn fats, and that capacity collapses with age.
In worms, the import machinery that arms peroxisomes, run by a protein called PEX5, declines first. Fat piles into droplets the cell cannot mobilize, and only then do the mitochondria swell and fail. The mitochondrial damage we treat as the cause looks downstream. What lands hardest is the dietary restriction result. It kept this machinery alive into old age, and switching it off erased the lifespan benefit entirely.
This is built almost entirely in worms, with little mammalian backup and none in humans. It is a mechanism, not a protocol.
Still, the shape of it is what matters. If metabolic aging runs in a sequence, what counts is knowing where you sit in it, and no standard checkup is built to tell you. 🧬
nature.com/articles/s43587-0…
2
42
May 20
Macular degeneration, liver fibrosis, AML. These are the diseases the new AI discovery systems chose to prove themselves on. Every one is a disease of aging.
The science is real. But the loop it speeds up is the one medicine has always run, chasing a disease after it surfaces, one at a time.
For a single person, the loop that actually compounds runs upstream of all of them. Read the trajectory while there's still room to change it, instead of waiting for the disease to show up. That's the layer almost no one is building for individuals. 🧬
May 19

A big day for multi-agent AI to accelerate biomedical discovery, hypothesis generation, designing experiments with proof points of new candidate drugs (cancer, fibrosis, macular degeneration, antimicrobial resistance, and more)
2 @Nature reports @GoogleDeepMind @FutureHouseSF
nature.com/articles/s41586-0…
nature.com/articles/s41586-0…
2
55
May 19
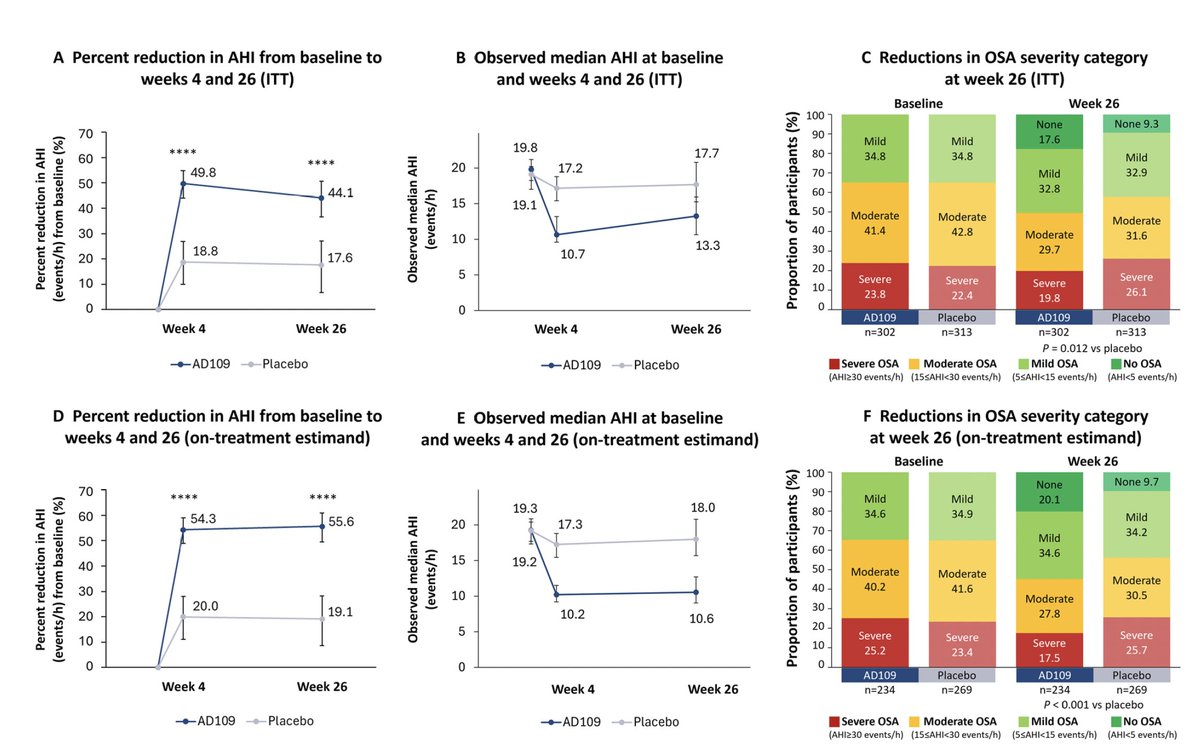
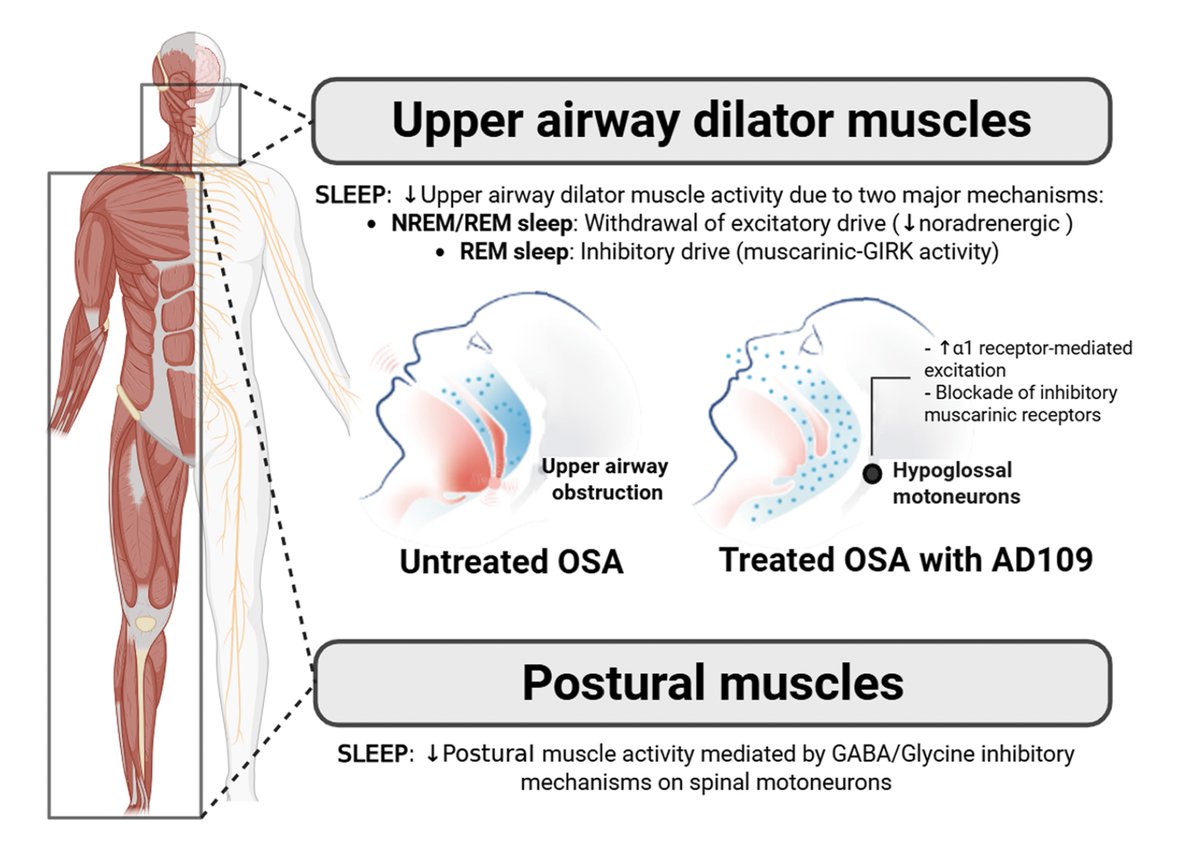
Apnimed's AD109 cleared Phase 3 on its primary endpoint. The patient-reported story is more interesting than the headline.
PROMIS-Fatigue and Sleep Impairment fell by 7.4 and 7.6 points on AD109, and by 6.2 and 6.9 on placebo. The drug-specific signal didn't reach ITT significance (p=0.059, p=0.316), though the on-treatment estimand for fatigue did (p=0.030), and patients with higher baseline fatigue or sleepiness clearly responded.
What this exposes is a measurement design issue more than a drug failure. A 6-month placebo-controlled snapshot can't cleanly isolate drug-specific signal when symptom self-report itself responds to being clinically watched.
A billion people have OSA, and eighty percent don't know it. The pharmacology is arriving. The infrastructure to track whether any of it actually moves a life trajectory across years isn't here yet.
May 18
A once a night pill for severe obstructive sleep apnea effective in a Phase 3 randomized trial
academic.oup.com/ajrccm/adva…
academic.oup.com/ajrcmb/adva…
1
2
78
May 14
Most high-performers run on a simple assumption, that more inputs mean better outputs. More sleep, more protein, more cold plunges, more NAD . Biology doesn't read its inputs that way. Almost every variable has a window, not a slope.
Nature this week added sleep to a long list. The MULTI Consortium analyzed 23 biological ageing clocks across imaging, plasma proteomics, and metabolomics in the UK Biobank. A consistent U-shape emerged across nine organ systems. The lowest biological age gaps land between 6.4 and 7.8 hours of sleep, with the optimum shifting by organ and by sex. Below 6 hours and above 8 hours both track to higher disease incidence and higher all-cause mortality.
The interesting question is no longer how much sleep. It's where your window is. Self-reported sleep is the methodological limitation here, so the 6.4-7.8 range is directional rather than prescriptive. The bigger pattern is robust. Your biology runs on dose-response. What's missing is the infrastructure to find your individual window. 🧬
1
1
3
78
May 14
45
May 13
The most oxygen-deprived tissue in your body ages the slowest. That is not a typo.
Your intervertebral disc lives in permanent hypoxia from birth. Most organs only run into low oxygen locally as they age, and when they do, a protein called HIF-1α builds up and accelerates cellular aging. The disc faces the same low-oxygen environment its entire life, yet keeps HIF-1α levels low and stable at every age. A team at Naval Medical University (Nature Aging, yesterday) showed why: the disc runs a selective autophagy pathway that continuously clears HIF-1α before it can accumulate. It solved a problem other organs never had to deal with until they started getting old.
The interesting part is what they did next. They built a small molecule called HATC that mimics the disc's clearing trick, gave it to aged mice weekly, and watched HIF-1α drop across multiple organs within two weeks. Median lifespan went up about 14 percent, maximum lifespan about 12 percent. These are mouse numbers. HIF-1α also does essential work in wound healing and blood vessel formation, so turning it down body-wide in humans is not straightforward. But the logic of the approach is what stays with you: find the tissue that ages slowest, reverse-engineer the mechanism, and export it outward. That is not how most drug discovery works. Maybe it should be.
1
2
60
May 13
How much does your DNA determine how long you live? Depends on who you ask and what exactly they measured.
Eric Topol sequenced the genomes of 1,400 people over 80 who never got a major chronic disease. He compared them with everyone else. The result was, in his words, "not much of a genetic story at all." Lifestyle, immune function, environment explained the gap. That was a study about healthspan, about whether you get sick.
Shenhar et al. published in Science this January asking a different question. Not who avoids disease, but how long people live. Using Swedish and Danish twin registries and stripping out deaths from accidents and infections, they landed on a heritability of about 50 percent. Twice the old consensus. But that number depends on a modeling choice about which deaths count as "biological aging" and which don't. A 2018 pedigree study controlling for assortative mating put the same number closer to 7 percent.
So the field has three answers to what sounds like one question: 7, 25, 50. They are not conflicting results. They are answers to three different questions that most people, and most health systems, have never been asked to tell apart. Whether you can avoid disease and how long your biology lets you live may not share the same levers at all. If that is true, then the 15-year gap between when healthspan ends and when lifespan ends is not one problem. It is two, with different drivers, and almost nothing in standard care is built to help you work on either one with any precision.
2
53
May 9
Your blood already contains a multi-organ aging map. A team from the Chinese Aging Biomarker Consortium just demonstrated this in Cell, building a three-tiered clock from 2,019 adults that reads six organ systems from plasma proteins alone. Your liver ages faster than your brain. That difference is quantifiable from a single draw.
What makes this more than another clock paper: the coagulation factors your doctor already measures for clotting risk are not just rising with age. They are driving it. The team treated human endothelial cells with these factors and got accelerated senescence. They injected them into mice and saw inflammatory damage across aorta, liver, heart, and kidneys. It opens a possibility that one of the most routine markers in medicine has been hiding an aging signal in plain sight.
The direction is clear: the science to read your body at organ-level resolution exists. The system to deliver that reading to you does not.
cell.com/cell/abstract/S0092…
1
61