
71 Photos and videos












Pinned Tweet

1 Jul 2024
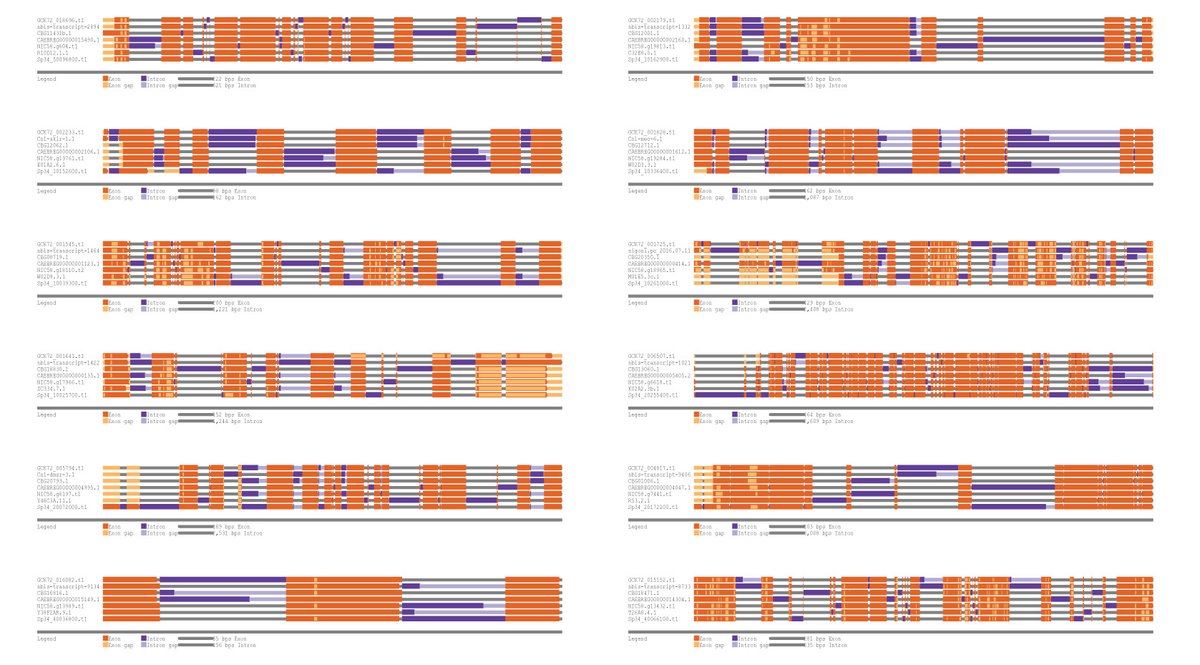
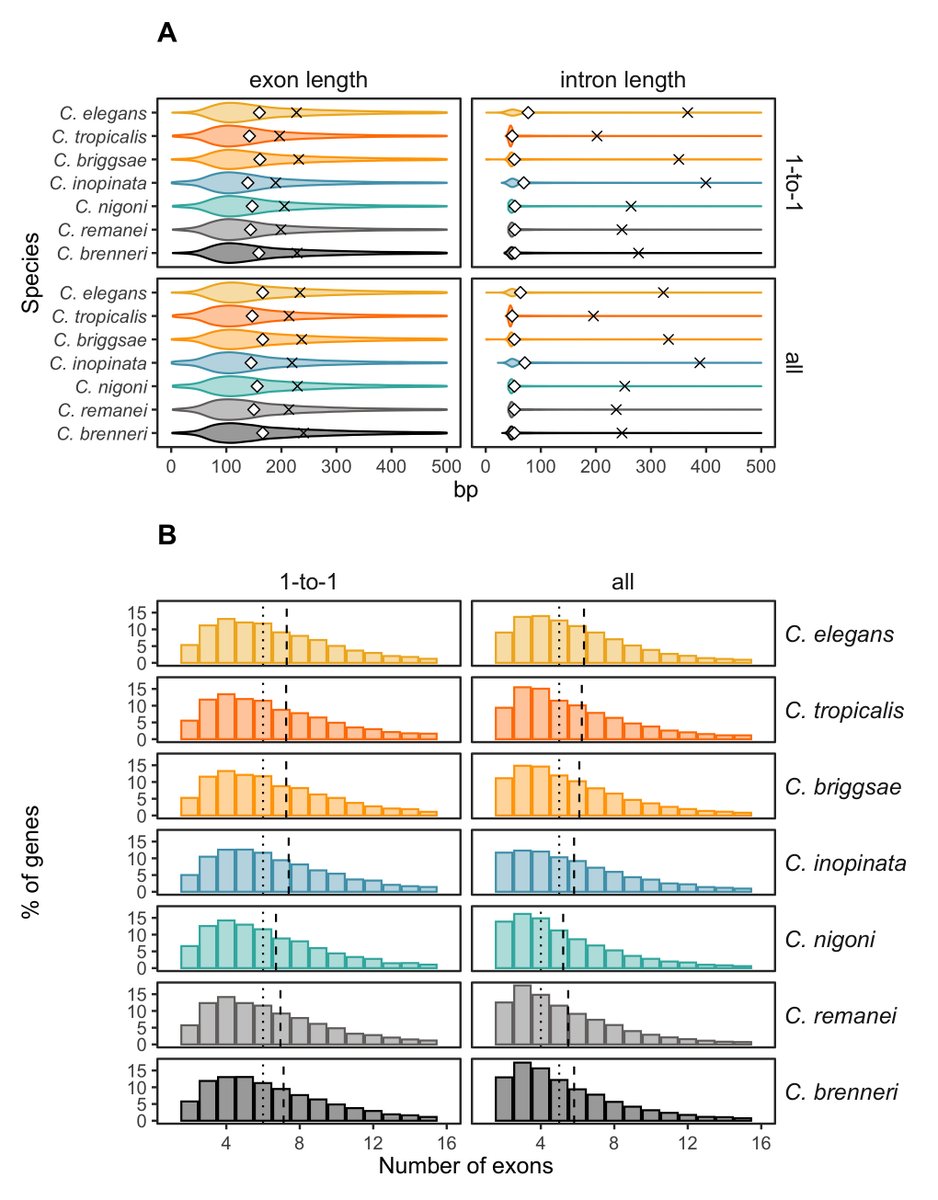
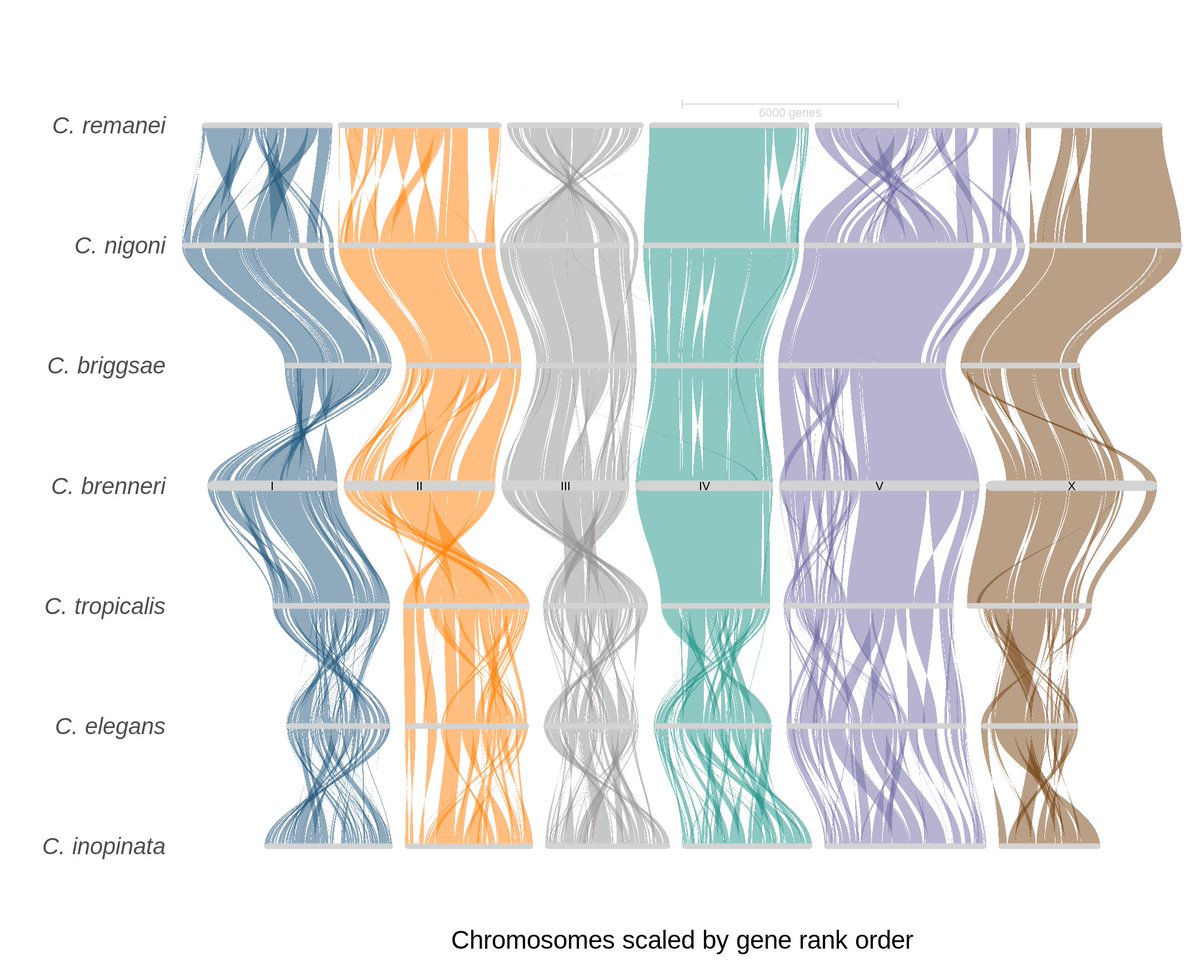
🚨 New preprint alert 🚨
We've generated a new gapless genome of Caenorhabditis brenneri🪱, one of the most diverse metazoans! We compared its genome and gene organizations with other nematodes with smaller population sizes. Check it out!
biorxiv.org/content/10.1101/…
1
7
18
1,864
Anastasia Teterina retweeted

🧬Collinearity Analysis is here in #gbdraw v0.11.0! You can easily detect and visualize synteny and collinearity blocks using LOSATP. You can instantly align the entire diagram based on a shared ortholog!
Try it now: gbdraw.app/
#bioinformatics #genomics #microbiology

ALT Collinearity analysis of Hepatoplasmataceae, a group of mollicutes associated with isopod crustaceans. Inverted syntenic blocks are colored in dark red.
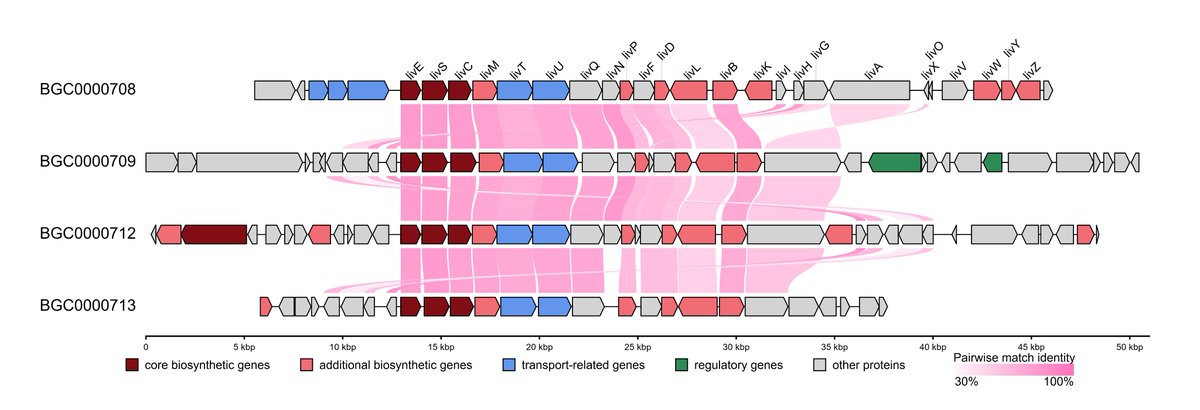
ALT Synteny of lividomycin-like aminoglycoside biosynthetic gene clusters. The diagrams are aligned based on livE gene (see next figure for the command)
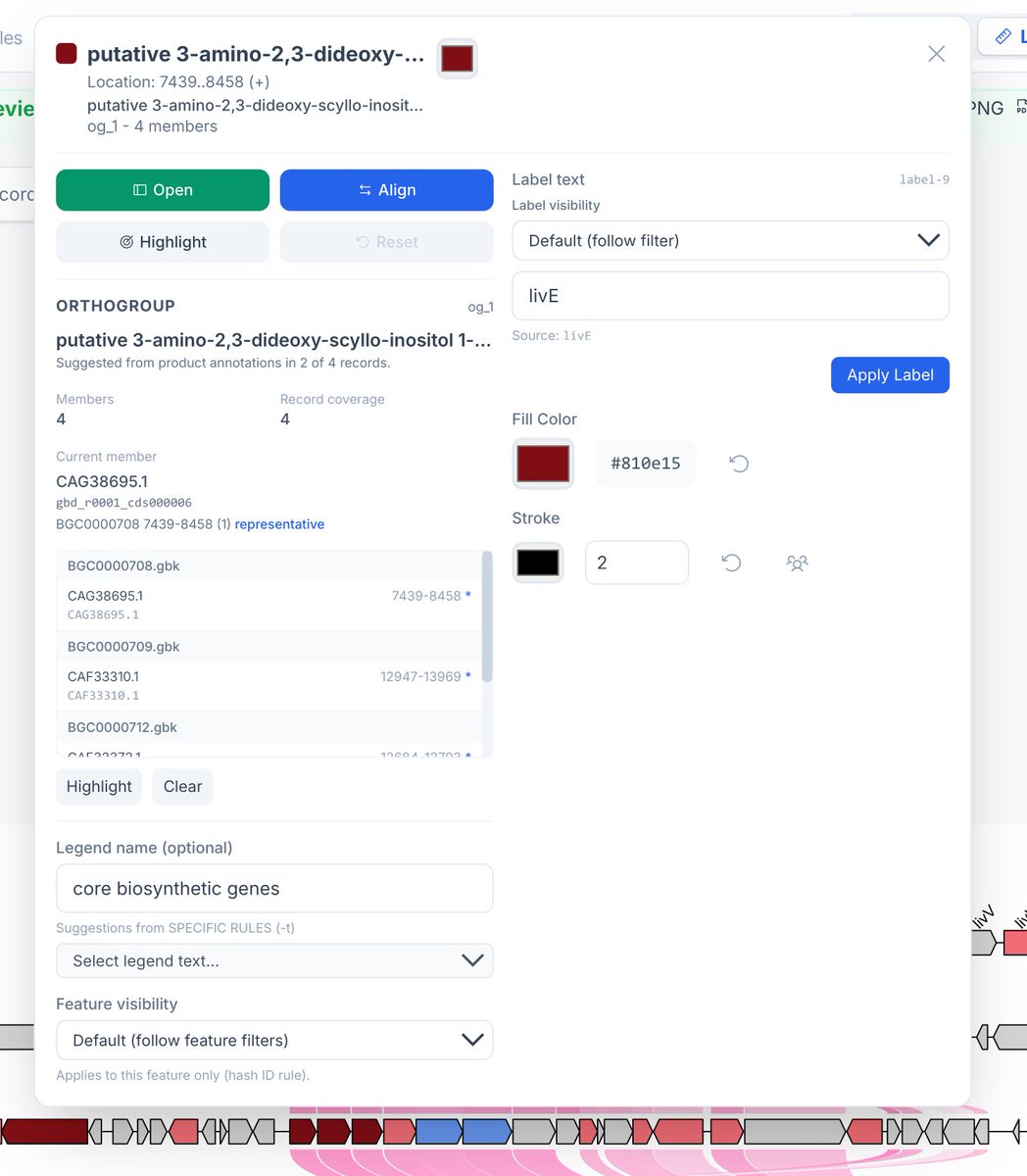
ALT Dialog box for the livE gene in the record BGC0000708. Click "Align" to align the records based on the shared livE orthologs, as seen in the previous figure.
1
49
171
9,728
Anastasia Teterina retweeted

25 Jun 2025
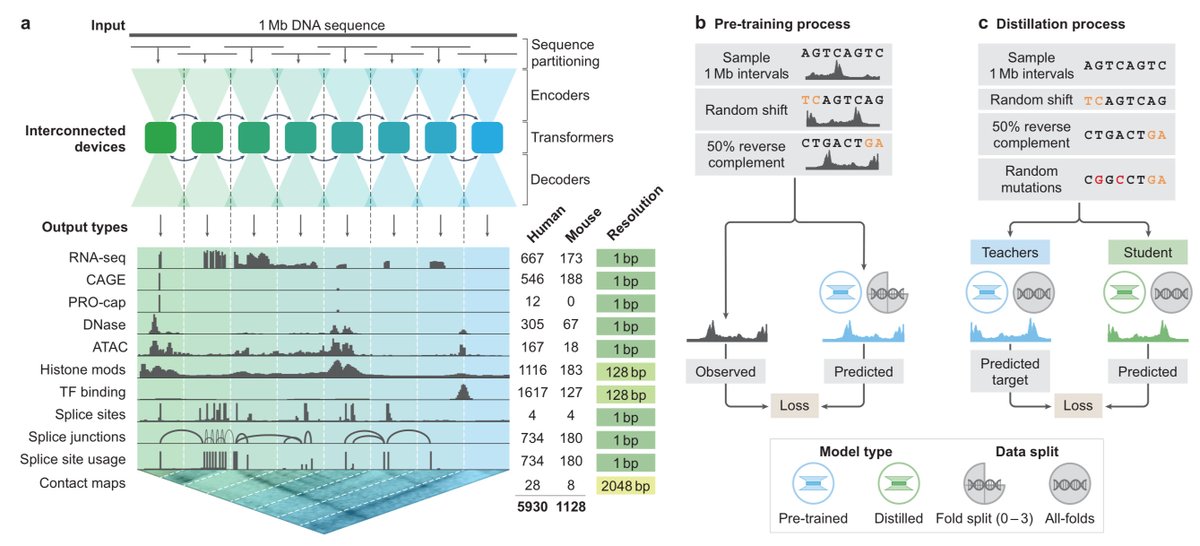
AlphaGenome: advancing regulatory variant effect prediction with a unified DNA sequence model storage.googleapis.com/deepm…
1
59
201
16,535
Anastasia Teterina retweeted
26 Sep 2024
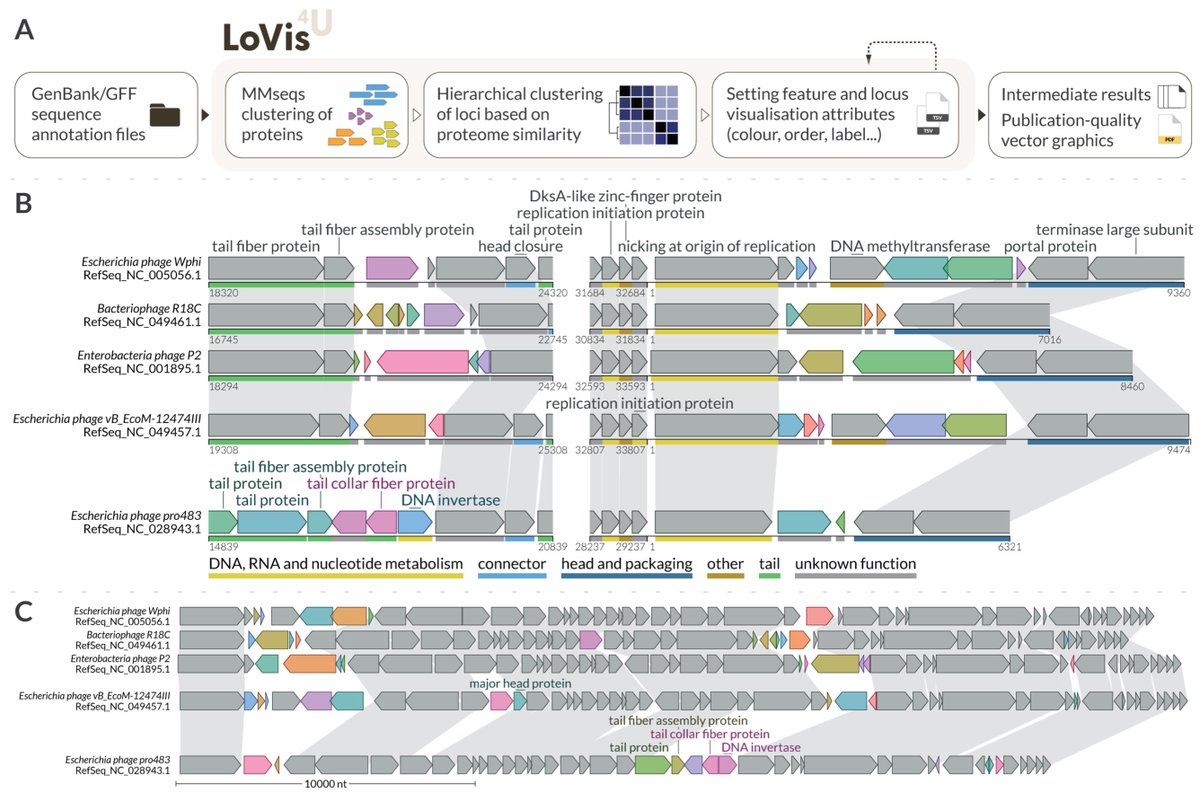
LoVis4u: Locus Visualisation tool for comparative genomics biorxiv.org/content/10.1101/…
1
73
261
21,344
Anastasia Teterina retweeted

29 Nov 2025
The model of gene expression taught in school is highly misleading!
Transcription factors are proteins that bind to DNA and then help repress, or activate, the expression of genes. Cells have hundreds of different types of transcription factors, each tuned to regulate different genes based on short snippets of DNA located near those genes.
The basic model, taught in school, says that these transcription factor proteins float around the cell and, when they bump into a DNA sequence, either latch onto it strongly (CORRECT SITE!) or fall off quickly (WRONG SITE) and keep searching. All the other DNA in a cell is basically abstracted away as unimportant or irrelevant; mere background noise.
But again, this model is naive! And a new paper, published in Cell, beautifully shows how the sequences SURROUNDING a transcription factor's binding site also matter a great deal.
This won't be surprising to many biologists, as "cracks" in the standard two-state model began emerging decades(?) ago. Biologists have tagged transcription factors with fluorescent tags and then watched them move around living cells. And they have noticed that when transcription factors land in a "wrong" location in the genome, they skip or hop to a nearby location and repeat this until finally connecting with the "correct" sequence. So in other words, there are actually three states that a transcription factor can exist in: free-floating, "searching", or "bound."
(More technically, transcription factors first do a 3D search, then latch onto DNA and do a 1D search to find the correct location.)
For this new paper, though, scientists exhaustively quantified *how* the sequences flanking a transcription factor binding site influence the search of the protein.
They did a huge in vitro experiment, wherein they placed a specific transcription factor with a known binding site, called KLF1, in a huge library of 11,812 different DNA sequences. These sequences had mutated "core" binding sites and variations in the flanking sequences. They also prepared negative controls. Then, these researchers measured the binding kinetics of KLF1 with each sequence to understand which bases in the flanking sites impact the 1D search.
What they found is that KLF1 has a basically flat disocciation rate from its core sequence, but that the PROBABILITY that it finds this sequence depends a lot on the surrounding context. Even mutations located dozens of bases away from the core site matter a lot, either pushing KLF1 to "hop" faster to find the site, or "trapping" KLF1 and slowing down its search. These flanking sequences can cause up to a 40-fold variation in the affinity of a transcription factor for its target site!
This is just one small part of the paper, though, so I encourage anyone interested to read the whole thing. It is challenging throughout.


18
237
1,178
127,119

I spent months illustrating how Transformers actually work.
Not just what they do, but why they’re built this way. The history, design choices, and intuition behind every layer.
From RNNs → Attention → Multi-Head → FFNs → Positional Encoding.
Here's everything I wish I knew:
71
372
3,266
248,710
Anastasia Teterina retweeted

29 Oct 2025
scooby: modeling multimodal genomic profiles from DNA sequence at single-cell resolution. #SingleCell #GeneExpression #Genomics #Transcriptomics #Epigenomics #Bioinformatics @naturemethods
nature.com/articles/s41592-0…

2
9
64
4,124
Anastasia Teterina retweeted
25 Oct 2025
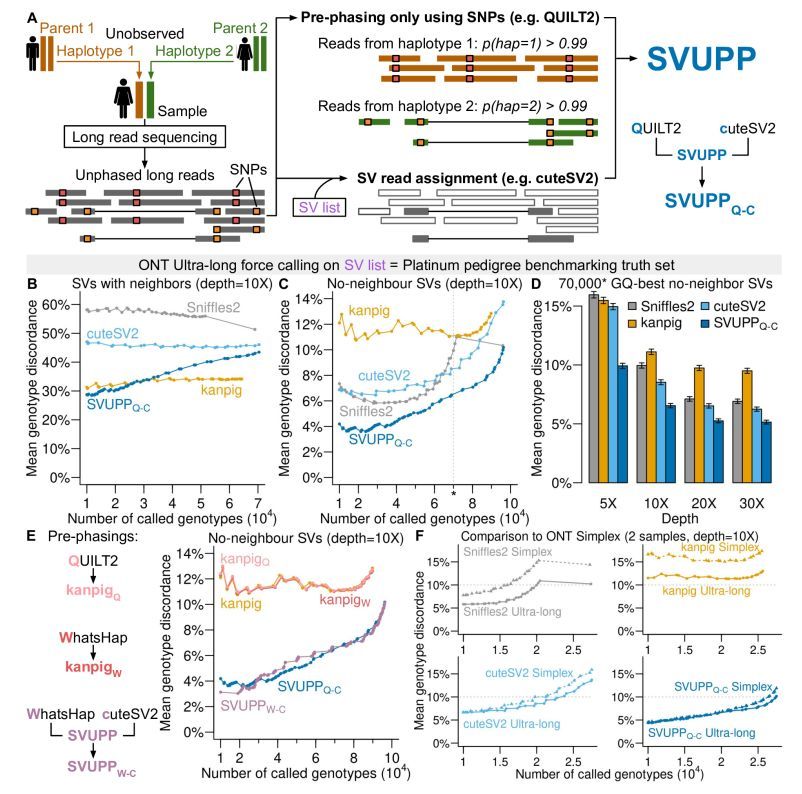
Pre-phasing long reads improves structural variant genotyping. #LongReads #Sequencing #ReadsPhasing #StructuralVariants #Genotyping #Genomics #Bioinformatics
academic.oup.com/bioinformat…
1
9
26
1,380
Anastasia Teterina retweeted
18 Oct 2025
GRiNS: a python library for simulating gene regulatory network dynamics bmcbioinformatics.biomedcent…

1
29
99
7,206
Anastasia Teterina retweeted
29 Aug 2024
OrthoBrowser: Gene Family Analysis and Visualization biorxiv.org/content/10.1101/…
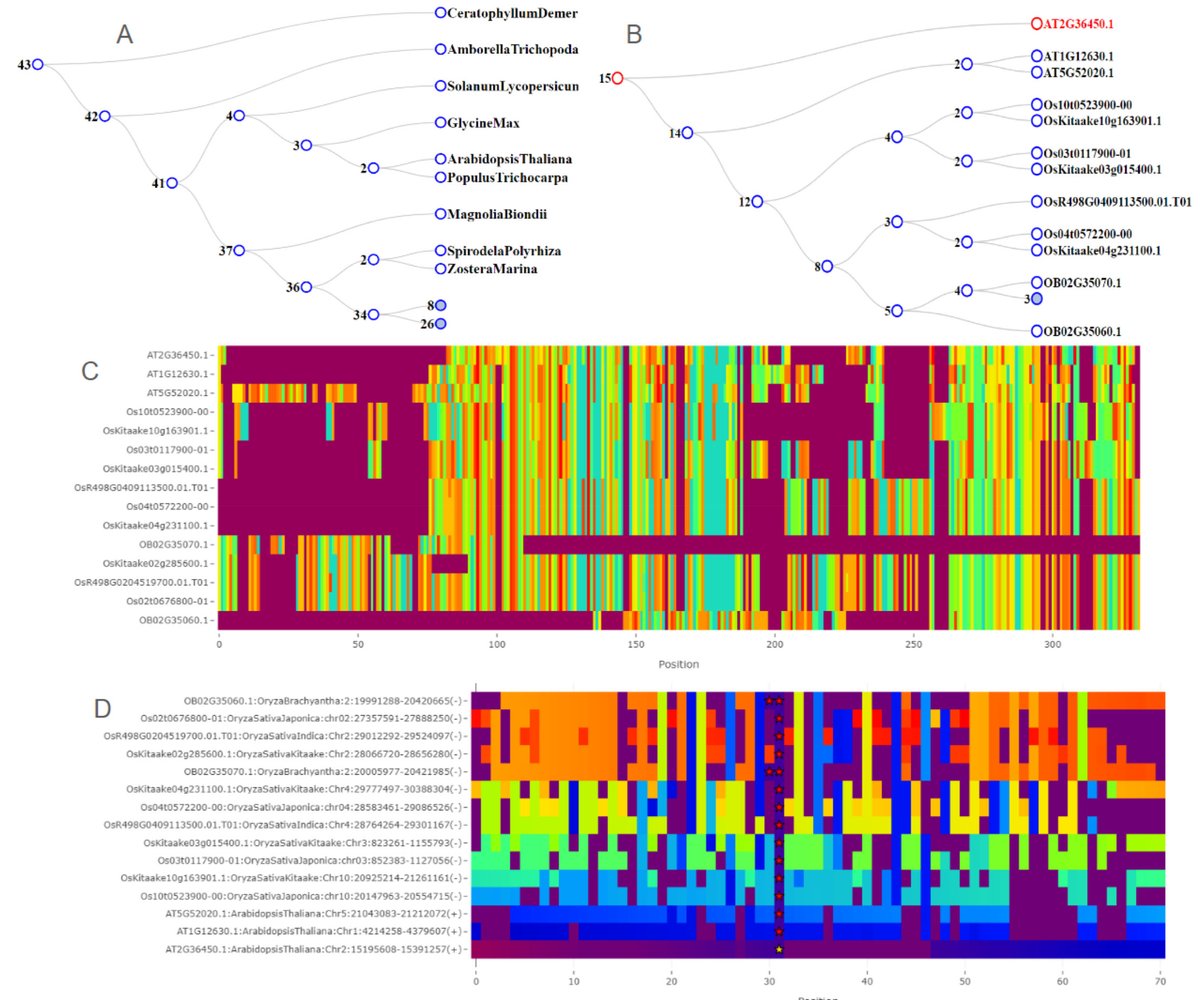
3
77
257
24,420
Anastasia Teterina retweeted
24 Oct 2024
G2PT: Genotype-to-Phenotype Transformer: Mechanistic genotype-phenotype translation using hierarchical transformers biorxiv.org/content/10.1101/…
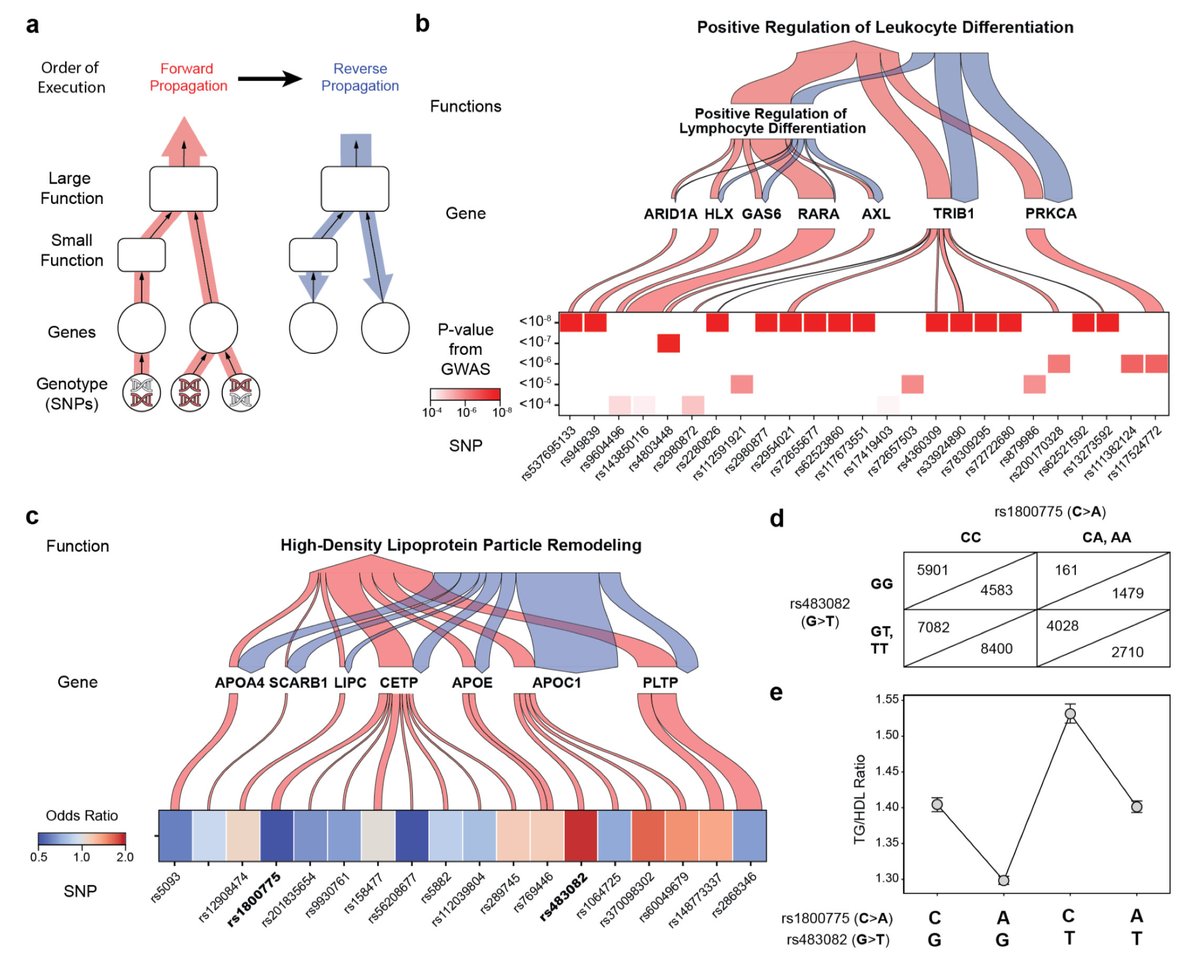
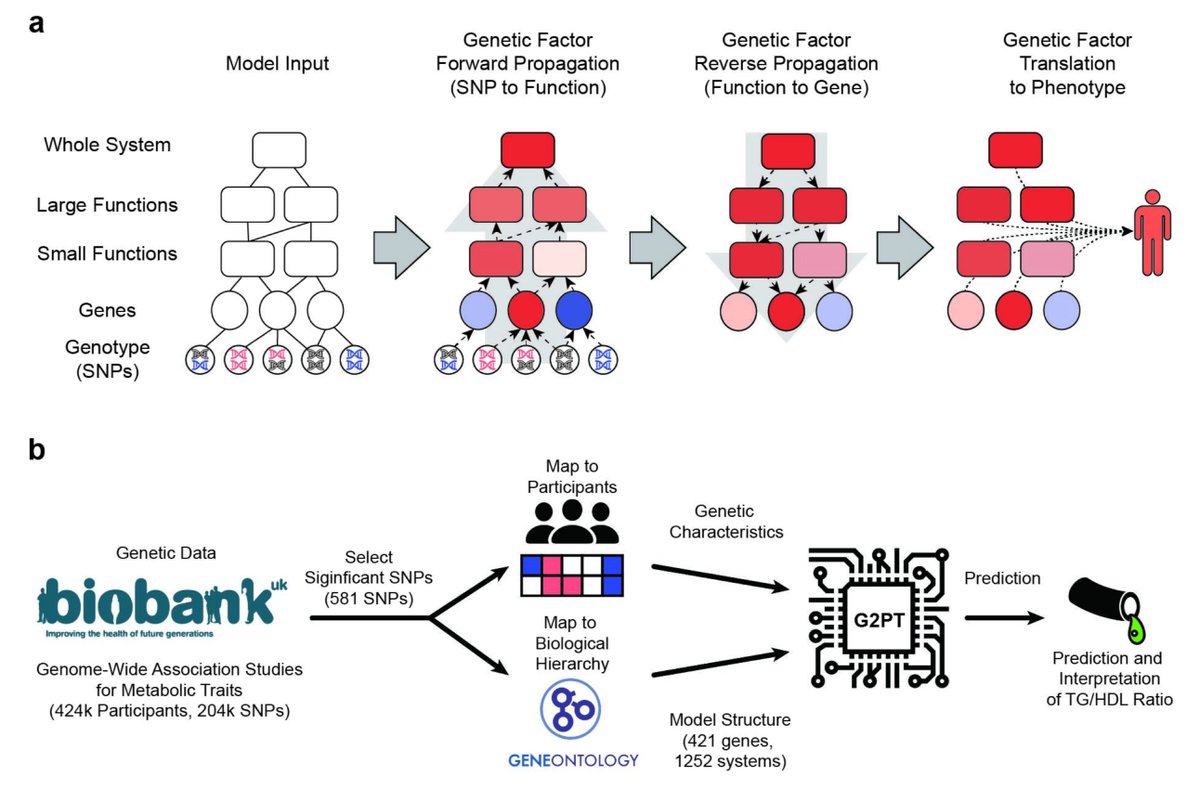
4
79
346
47,167
Anastasia Teterina retweeted
12 Jun 2025
DiSC: a Statistical Tool for Fast Differential Expression Analysis of Individual-level Single-cell RNA-seq Data.#SingleCell #RNAseq #scRNAseq #DifferentialExpression #Bioinformatics
academic.oup.com/bioinformat…
1
6
16
740
Anastasia Teterina retweeted
7 Jun 2025
Pangenome-aware DeepVariant biorxiv.org/content/10.1101/…
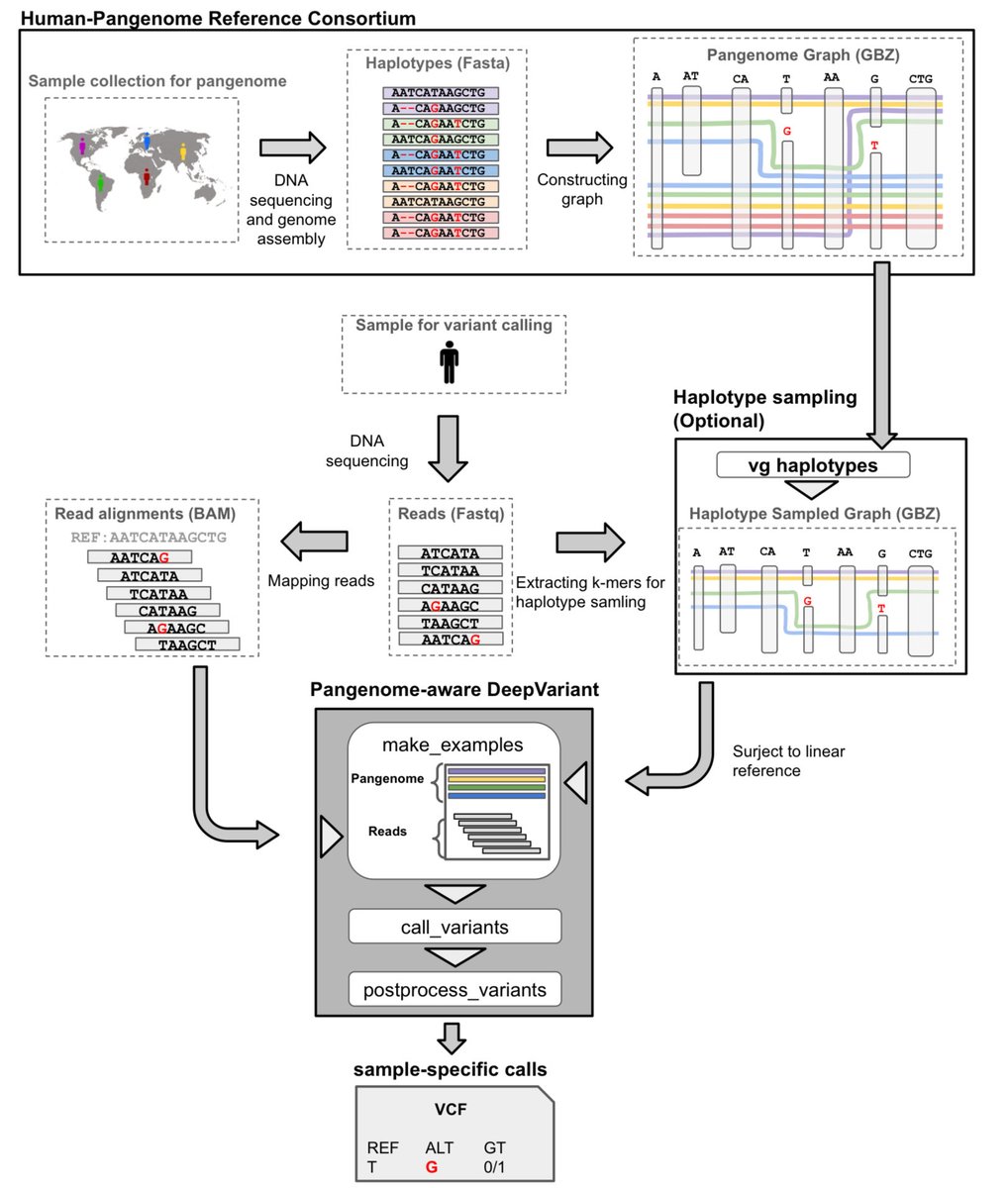
1
22
93
5,734
Anastasia Teterina retweeted

17 Jan 2025
ntSynt-viz: Visualizing synteny patterns across multiple genomes biorxiv.org/cgi/content/shor… #biorxiv_genomic
6
16
4,186
Anastasia Teterina retweeted
19 Nov 2024
sTELLeR: Detecting transposable elements in long read genomes academic.oup.com/bioinformat…
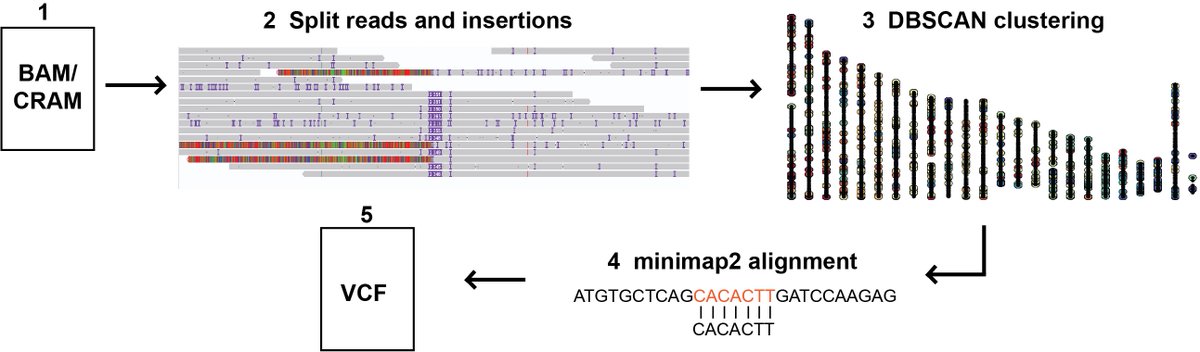
1
43
122
12,735
Anastasia Teterina retweeted

18 Nov 2024
A gentle introduction to pangenomics academic.oup.com/bib/article…
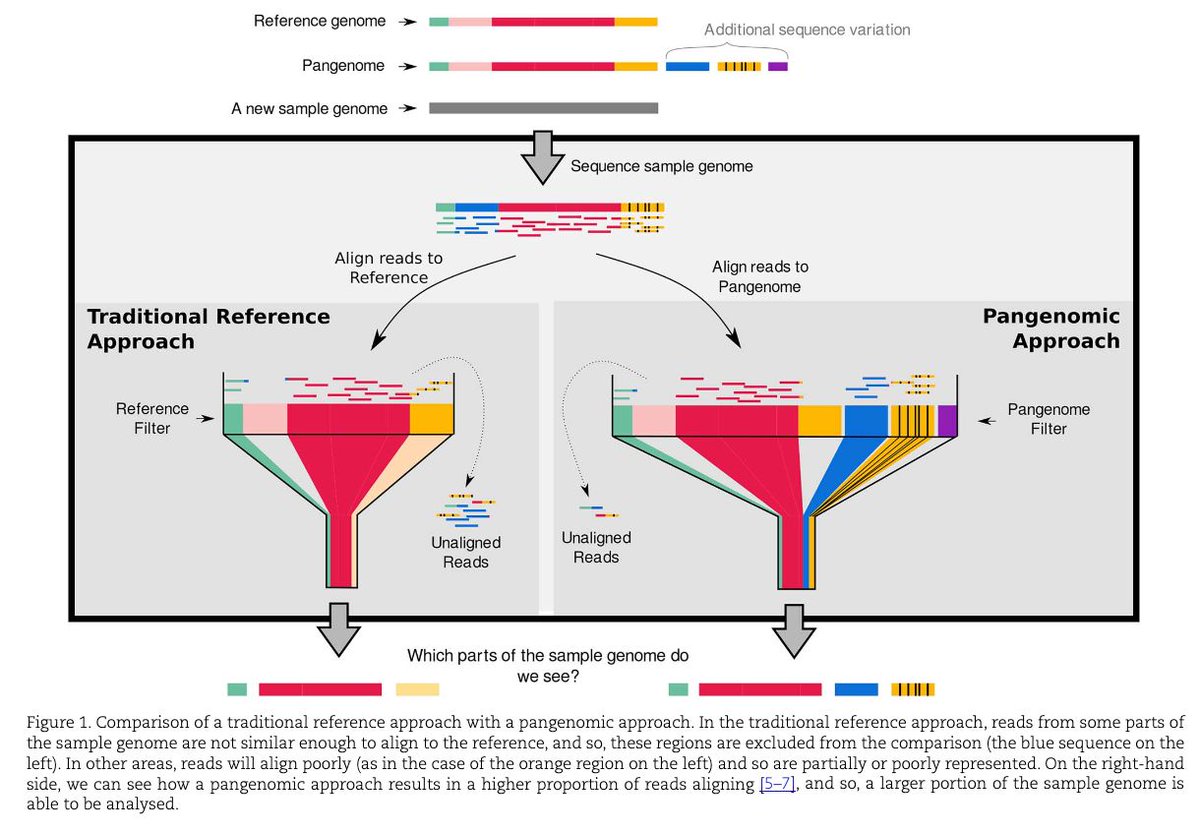
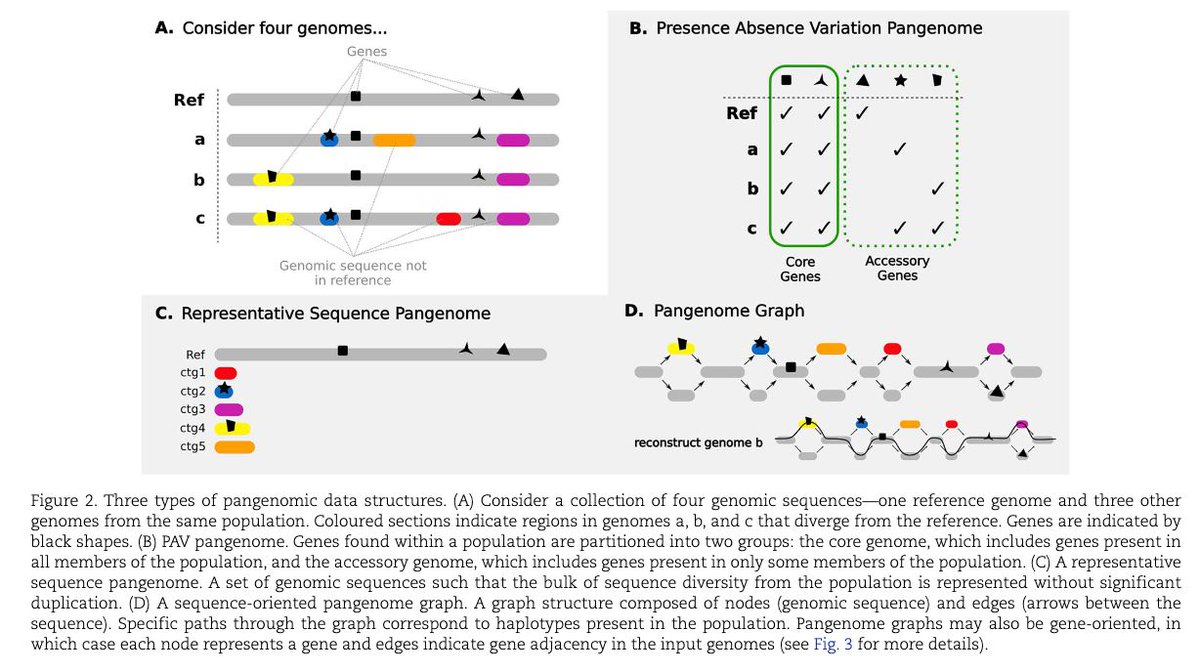
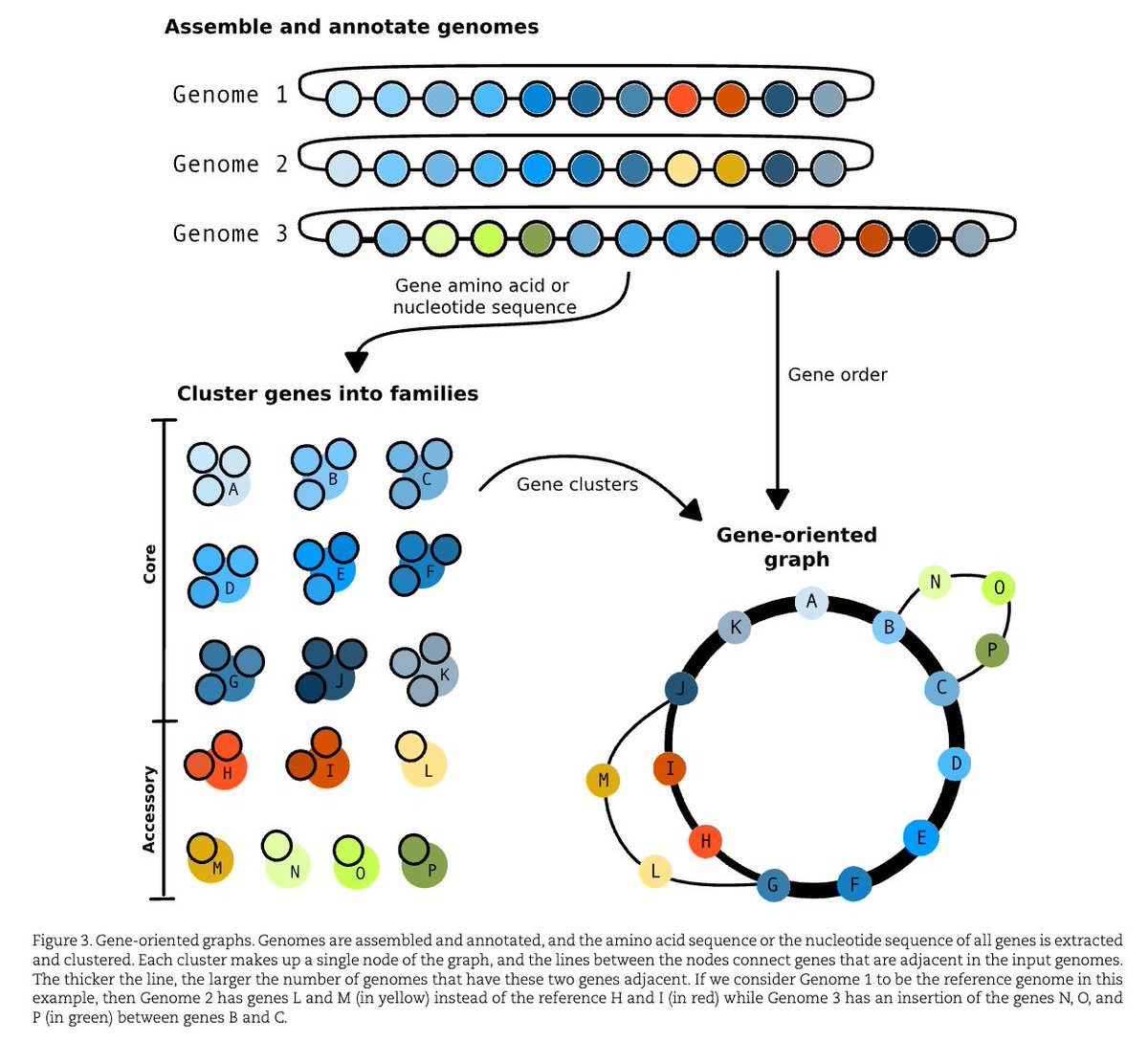
1
82
283
19,531
Anastasia Teterina retweeted

17 Oct 2024
REPORTH: Determining orthologous locations of repetitive sequences between genomes biorxiv.org/cgi/content/shor… #biorxiv_bioinfo
5
10
1,404
Anastasia Teterina retweeted

20 Sep 2024
🚨 Excited to share this #preprint from my postdoc in the Lippman lab @CSHL, in collab. with @mike_schatz & many others! Using pan-genomics & pan-genetics across the Solanum genus 🍅🥔🍆 we reveal gene duplications 🧬 as contingencies in crop engineering. doi.org/10.1101/2024.09.10.6…
7
44
135
10,880
Anastasia Teterina retweeted

24 Sep 2024
Widely observed negative correlation between recombination rate and TE abundance is usually interpreted as recombination rate being the "cause," influencing the efficacy of selection purging TEs. Yet, our recent study "flipped" this causality! biorxiv.org/content/10.1101/…… (1/5)
5
46
124
17,747
Anastasia Teterina retweeted

23 Sep 2024
Have you ever wondered whether modularity is the only driver of transcriptional network organization? Short answer, no! There are nuances to transcriptional networks, and together with @diogro and Julien Ayroles we explored some methods to find them: journals.plos.org/ploscompbi…
6
28
2,964
Anastasia Teterina retweeted

6 Sep 2024
Advertising this amazing tutorial that Rob Lanfear (no longer on this site) put together:
"For phylo nerds interested in concordance and discordance: iqtree.org/doc/recipes/conco…"
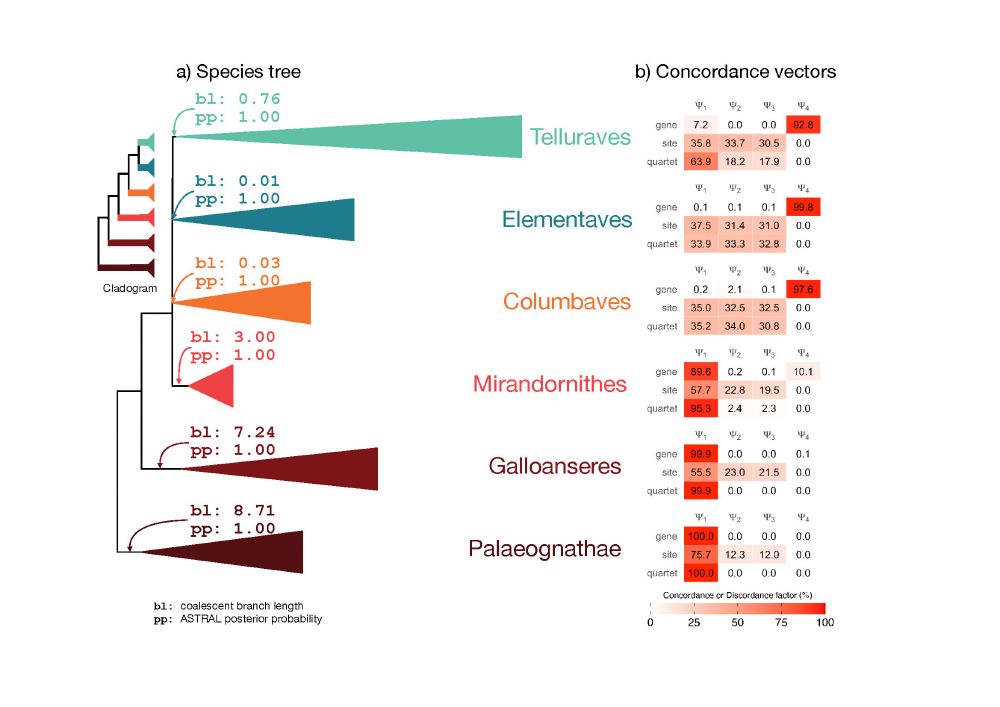
ALT Figure of a species tree with branch lengths and posterior probabilities (left) and a table of concordance vectors (right)
1
46
122
22,254