
ENCODE DCC * IGVF DACC (he/him)
Joined July 2010
- Tweets 1,152
- Following 407
- Followers 121
- Likes 1,440
21 Photos and videos














Pinned Tweet

21 May 2021
This is an infrastructure project.
Join us gopetition.com/petitions/a-u…
@anshulkundaje @ontowonka 16/16
3
4
Ben Hitz retweeted

26 Aug 2024
1/ One of the most common mistakes in bioinformatics is the one-off error. If you are not careful, you will make mistakes. Even experienced bioinformaticians are not aware of it and the mistake prevails in many bioinformatics tools. A thread 🧵
2
44
173
27,084
Ben Hitz retweeted

15 May 2024
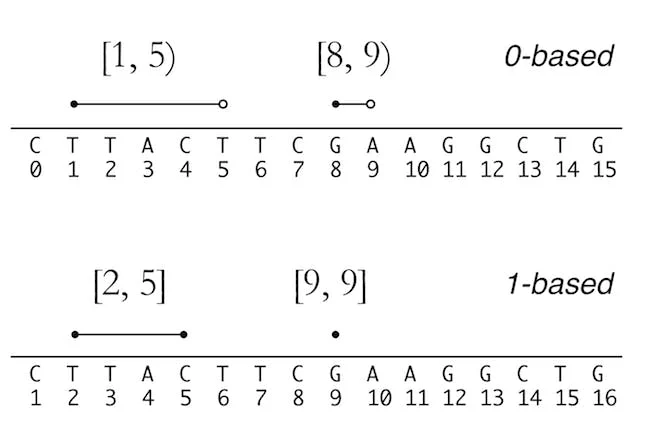
One funny story about this: I spent hours creating a figure in my book explaining 0 versus 1-based indexing and closed versus right-open intervals. The illustrator thought I made a careless error in starting one from 0 and the other from 1, and changed them to match 😱
15 May 2024

One of the many terrifying things I learned from your Bioinformatics Data Skills book!

2
35
4,671
Ben Hitz retweeted

10 May 2024
Dear big consortia,
It is never too late to be brave and use all that visibility you have to make a strong statement that academia will not be held hostage by glam journals & their shiny JIFs 1/
3
14
99
29,313
Ben Hitz retweeted

18 Apr 2024
Our paper "A machine readable specification for genomics assays" is now published in Bioinformatics, @OUPBioinfo. In short, we present a lightweight file format and command-line tool to document the structure of sequencing reads. Coauthored with @XiChenUoM and @lpachter.
Paper: doi.org/10.1093/bioinformati…
Code: github.com/pachterlab/seqspe…
What is in my sequencing reads?
Sequencing machines produce text files, called FASTQs, that contain reads or sequences of DNA molecules. Assay developers and data generators deeply understand the contents of their reads; they know the location and presence biological and synthetic constructs like cellular barcodes. Collaborators, reproducers, and other scientists may not despite their sometimes obscure addition to supplementary material.
Take for example the @10xGenomics Multiome assay. The 10x Genomics documentation [1] spells out the read structure for each modality: RNA reads contain a synthetic 16bp barcode, a 12 bp "randomly" generated unique molecular barcode (UMI), as well as cDNA that was captured via polyA capture. The ATAC reads consist of genomic DNA and a 16bp cellular barcode. However the 10x Genomics website explains that the ATAC portion of the 10x Multiome data contains an little-known 8bp constant sequence spacer that proceeds the 16bp cell barcode. So saying that you have "10x Multiome" reads is a necessary but not sufficient condition to know the contents of your FASTQ reads.
The reason is because the FASTQ read structure is dependent on both the assay as well as the sequencing machine/recipe used; a sequencing library produced from one single-cell assay can yield different read structures depending these parameters. Take the the 10x Multiome assay. The ATAC 24bp barcode spacer is usually sequenced as the i5 index read. Since the NextSeq 500/550 does not support a 24bp i5 read, the user must specify "dark cycles" (10x details the impact of this [2]) to skip the 8bp spacer. This yields a 16bp cell barcode in the i5 FASTQ file. If, however, the 10x Multiome ATAC library was not sequenced with dark cycles then the i5 FASTQ file will contain a 24bp spacer barcode. I was originally unaware of the 8bp spacer and the use of dark cycles in Multiome library sequencing. But as I was recently looking ATAC reads I realized the impact it had on my count matrices; I had been extracting half of the cell barcode and all of the spacer. This meant I was performing barcode error correction and was UMI collapsing the, mostly similar, cell barcodes to produce few counts.
This decoupling of read structure between the sequencing machine and the assay places a high priority on documenting read structure in a sequencer and assay-specific manner so that preprocessing tools can accurately extract and process relevant sequenced elements.
A machine-readable specification
I was inspired by @XiChenUoM 's efforts (which started while in @teichlab) in documenting sequencing reads of assays and I came up with an idea to document read structure in a machine- and human-readable specification.
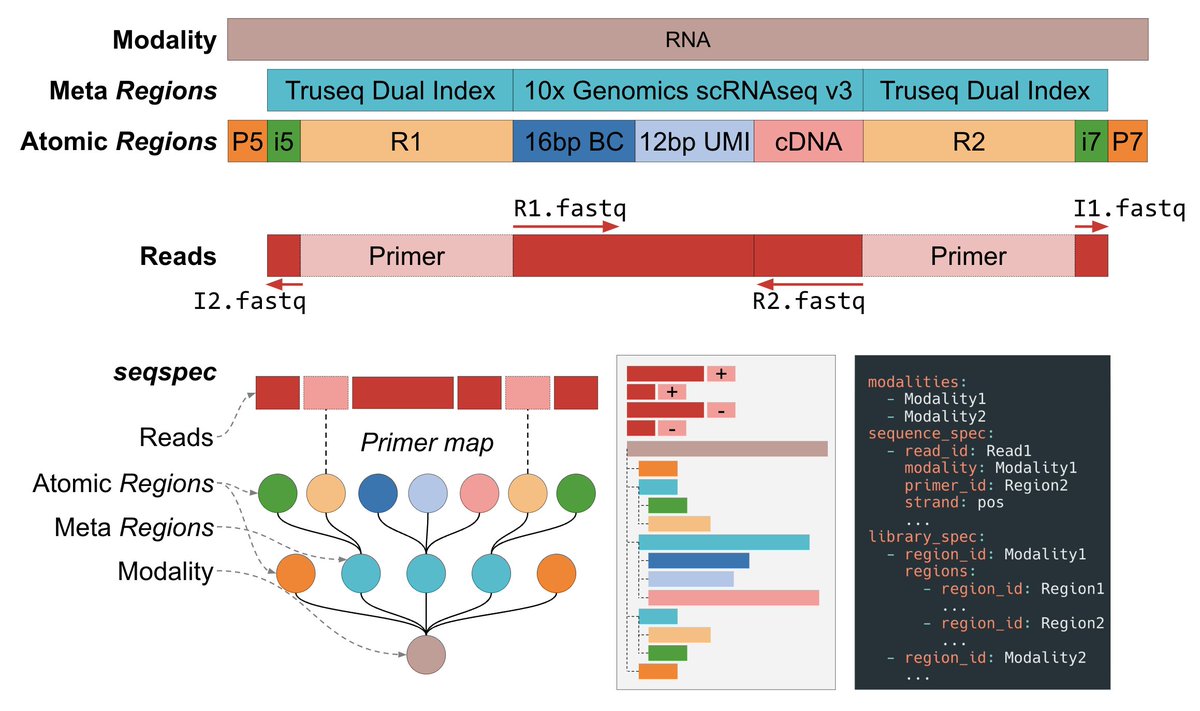
The specification is called seqspec [3]. The specification details the structure of a YAML file that allows users to specify and annotate the types of sequences that are contained in their FASTQ data. seqspec uses a nested representation of "Regions" and "Reads" that allows users to annotate groups of sequenced elements and map sequencing reads to sequencing primers. This enables, for example, all of the elements contained in Read 1 of a FASTQ file, such as the barcode and UMI in the 10x RNA assay to be annotated as belonging to Read 1. The spec also comes with an accompanying seqspec command line tool which gives users who annotate their sequencing assays many benefits:
1. Reproducibility and verifiability of the assay structure
2. Positional extraction of relevant features
3. Visualization of the sequencing structure
The seqspec command line tool makes it straightforward to extract the positional index of sequenced elements. The barcodes in the 10x Multiome dataset could have easily been identified as starting 8bp into the reads with the seqspec index command. The tool also also makes it straightforward to visualize the structure of your sequencing reads. seqspec print can produce publication-ready figure of your read structure. Most importantly, seqspec makes it easy for others to reanalyze data for which a seqspec exists, bringing about verifiability of analysis results.
seqspec adoption
seqspec aims to make genomics processing correct and reproducible. seqspec was recently adopted as the first standard in the @IGVFConsortium and we anticipate the publication of terabytes of sequencing data alongside their seqspec read annotations. I personally believe seqspec will be transformative for reproducibility and analysis efforts, in particular for those undertaken by consortia. I hope that public databases (like the @NCBI SRA/GEO and DDBJ) will test out seqspec and look to adopt it as a standard for data submission.
seqspec is freely available, open source, open to contributions, useable, and well documented. Please take a look at the GitHub repo and try it out! We welcome feedback.
[1] 10xgenomics.com/support/sing…
[2] kb.10xgenomics.com/hc/en-us/…
[3] github.com/pachterlab/seqspe…
38
131
18,970
Ben Hitz retweeted
17 Jan 2024
I think the sad actual answer to "What is the biggest open challenge in biology?" is:
1. Getting people to share data.
2. Structuring, organizing, and annotating data with metadata so it's useful.
3. Building higher-level abstractions so people can efficiently work with big data.
23
73
376
122,912
Ben Hitz retweeted

29 Jun 2022
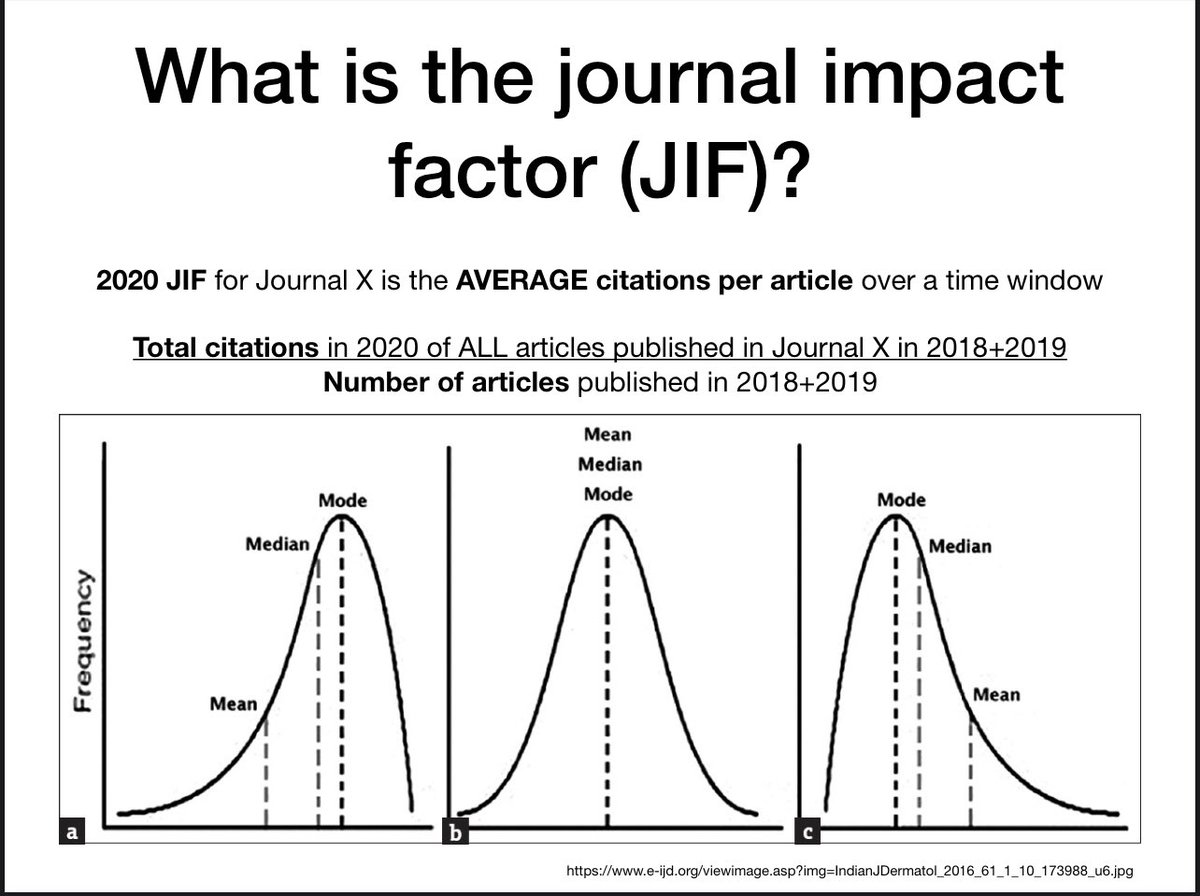
Here are some slides I show sometimes with data (from biorxiv.org/content/10.1101/…) on skewed journal citation distributions. JIF is a preposterous and inappropriate measure of individual article quality. Don’t get beclowned by the JIF
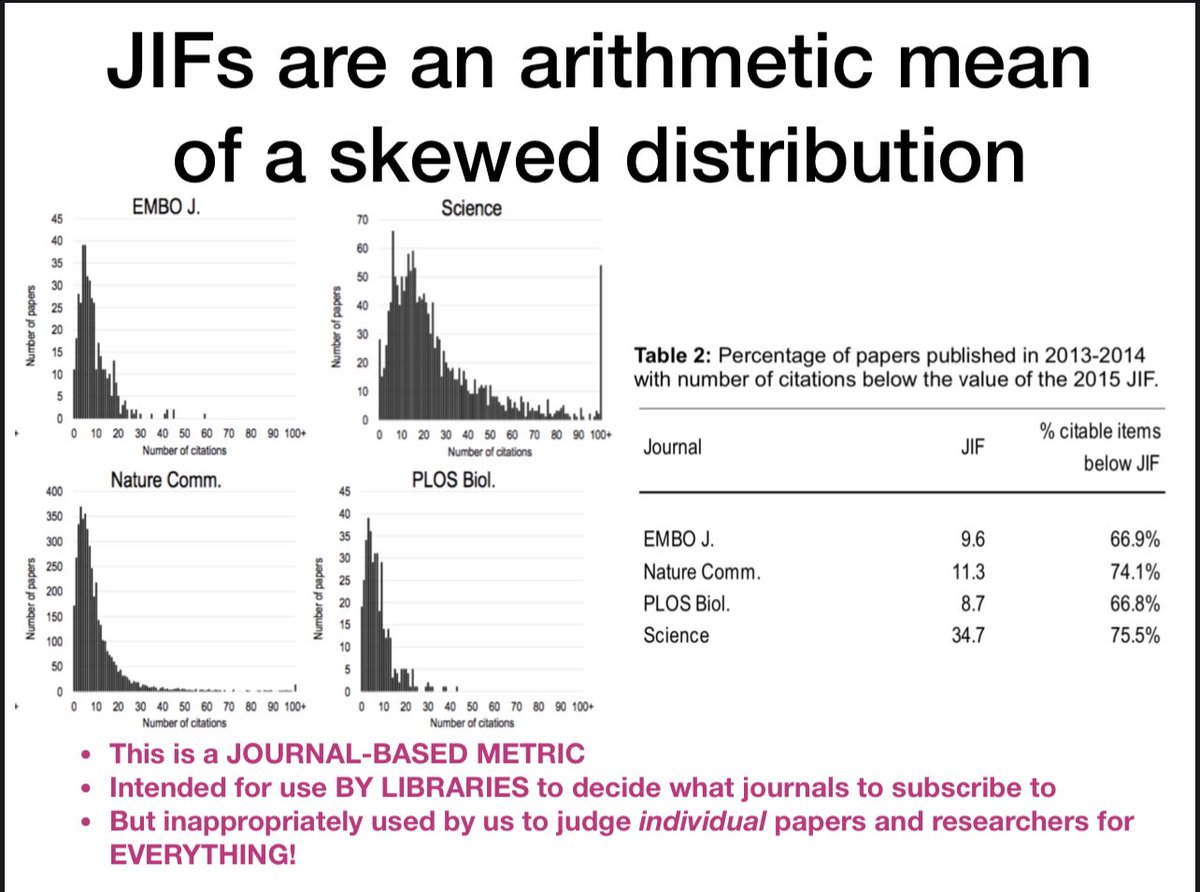

13
195
580
Ben Hitz retweeted

14 Nov 2023
ENCODE-rE2G: An encyclopedia of enhancer-gene regulatory interactions in the human genome biorxiv.org/content/10.1101/…
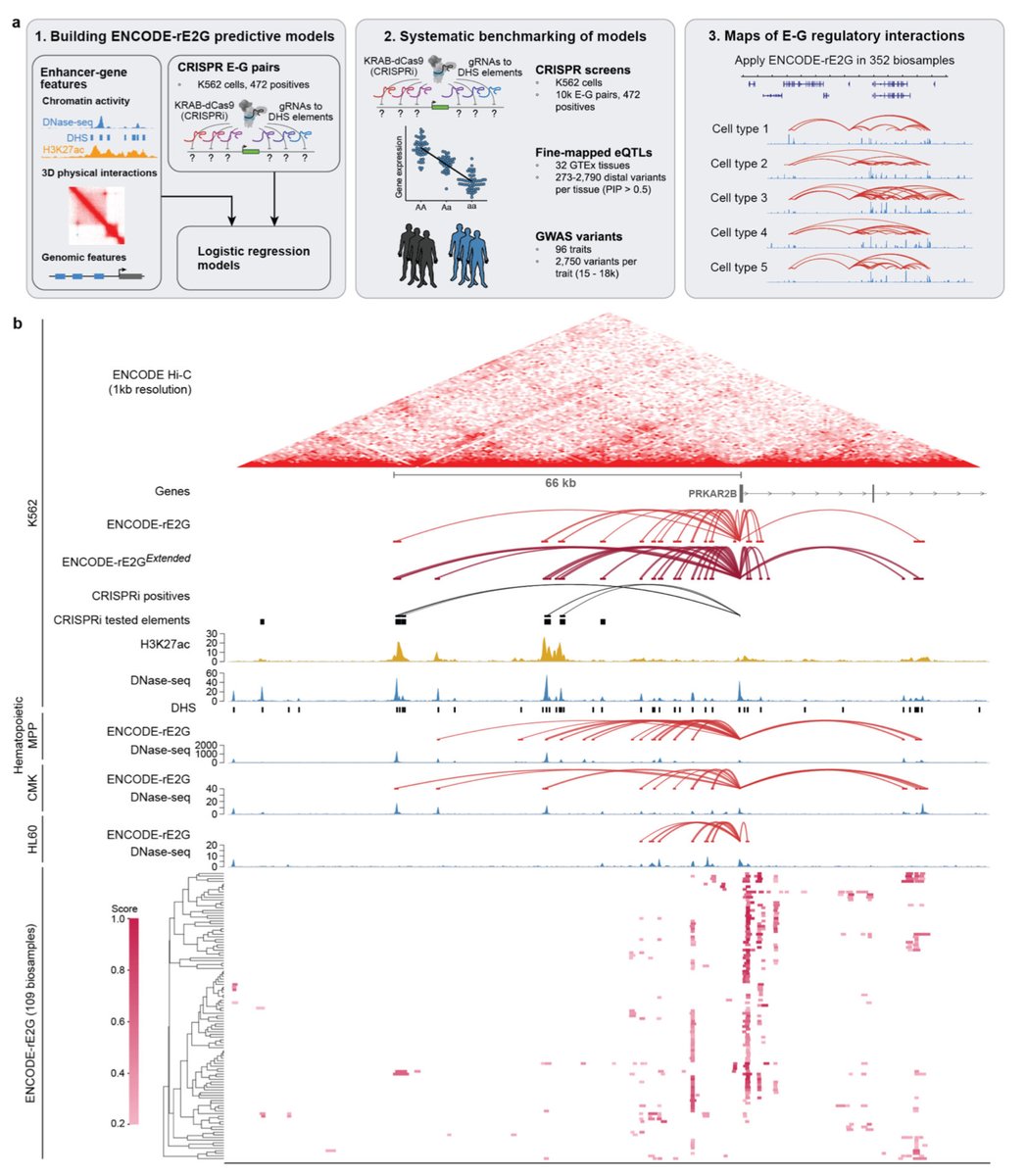
46
176
24,327
Ben Hitz retweeted

14 Nov 2023
Excited to share our latest work on bioRxiv!
biorxiv.org/content/10.1101/…
A highly collaborative effort with @ksmualim @karbalayghareh, @mayayayas, @kanishkadey, @evelynjagoda,@WangXijhu, @LarsMSteinmetz, @anshulkundaje, @jengreitz and many others from @ENCODE_NIH!
Thread👇
3
101
328
138,761
Ben Hitz retweeted

14 Nov 2023
Excited to share this ENCODE collaboration on the latest enhancer-gene regulatory maps to link GWAS variants to target genes and infer likely causal cell types.
14 Nov 2023

Excited to share our latest work on bioRxiv!
biorxiv.org/content/10.1101/…
A highly collaborative effort with @ksmualim @karbalayghareh, @mayayayas, @kanishkadey, @evelynjagoda,@WangXijhu, @LarsMSteinmetz, @anshulkundaje, @jengreitz and many others from @ENCODE_NIH!
Thread👇
1
9
32
8,369
Ben Hitz retweeted

7 Nov 2023
New preprint: "Integrative chromatin state annotation of 234 human ENCODE4 cell types using Segway reveals disease drivers" @marjanfarahbod @mforoozz @HDaneshpajouh @bonscotthoughts @JMichaelCherry1 @maxlibbrecht biorxiv.org/content/10.1101/…
1
8
12
2,028
Ben Hitz retweeted

16 Aug 2023
This paper is very worth reading for anyone interested in science, or who’s ever told something by a doctor. Inference uncertainty is very easy to confuse with outcome variability, and it’s easy to mislead readers and practitioners
(h/t @CT_Bergstrom)
pnas.org/doi/full/10.1073/pn…
6
74
252
50,110
Ben Hitz retweeted

30 Jul 2023
The Impact of Genomic Variation on Function Consortium (@IGVFConsortium) has published a "marker" preprint on the @arxiv: arxiv.org/abs/2307.13708
21
76
16,161
Ben Hitz retweeted

15 Jul 2023
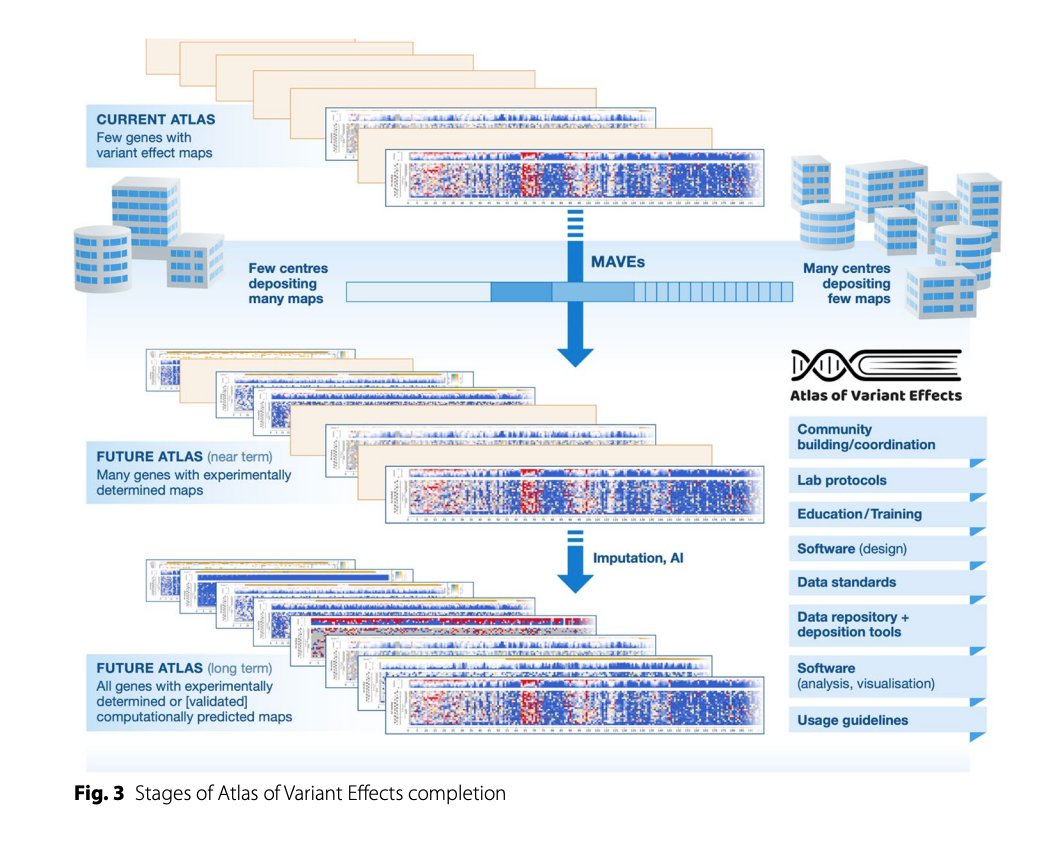
An Atlas of Variant Effects to understand the genome at nucleotide resolution | Genome Biology
genomebiology.biomedcentral.…
#genomics
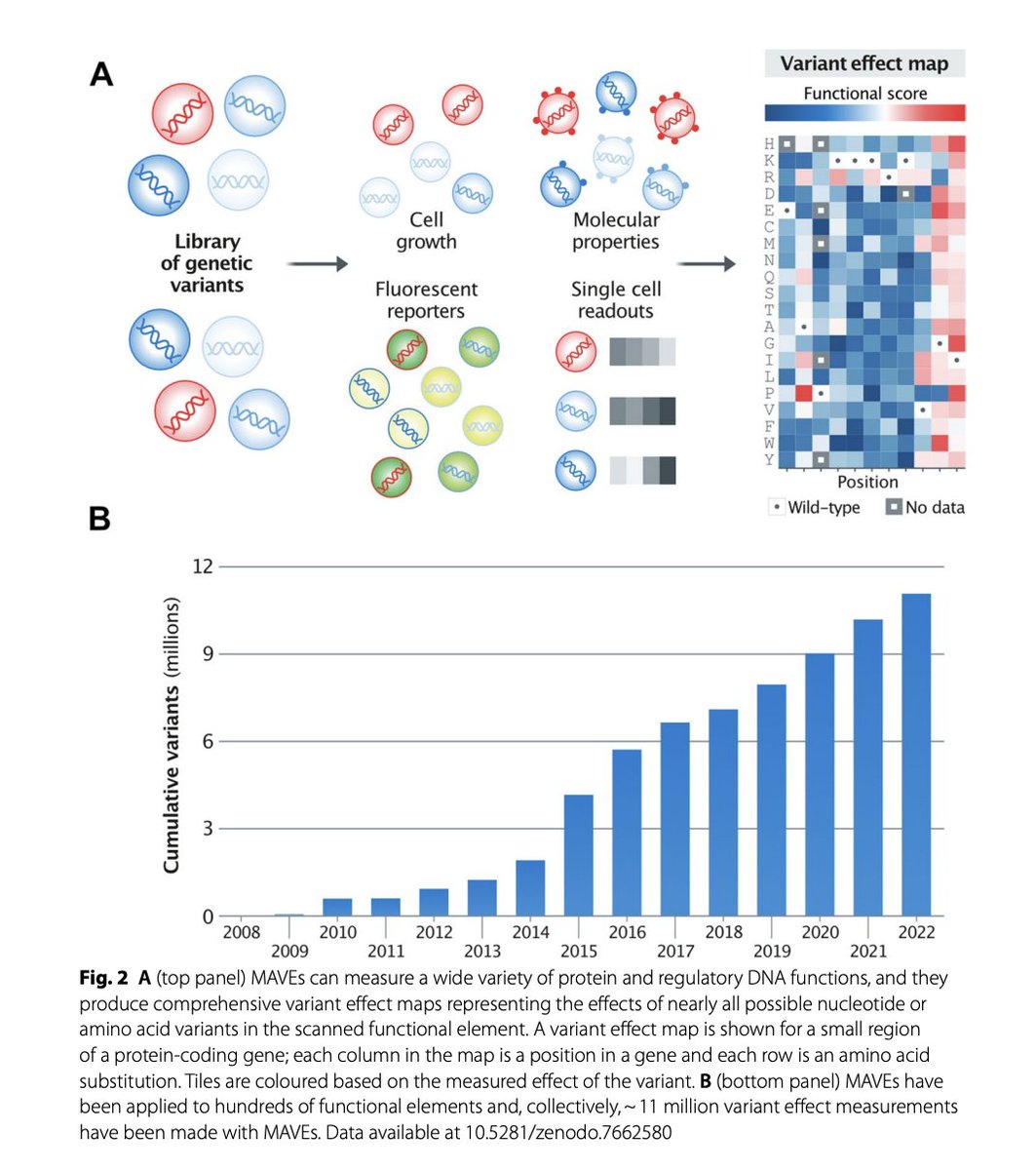
6
21
2,499

We are deeply saddened by the loss of Michael Ashburner, a pioneering co-founder and former Head of Research at EMBL-EBI.
His contributions to bioinformatics have been immeasurable. Our thoughts are with his family, colleagues and all those whose lives were enriched by his work.

ALT man leaning against a sign reading "EMBL Outstation Hinxton"
18
116
415
120,993
26 Jun 2023
Why not just publish the data and skip the paper?
23 Jun 2023

Introducing "data-to-paper": autonomous AI research! We've let it play with the large CDC Health Survey Dataset. Went to lunch. When back, it had already chosen several research topics, wrote data analysis codes, interpreted results and wrote 5 transparent, reproducible papers.

1
5
124
Ben Hitz retweeted

21 May 2023
Imagine writing this paragraph about our dysfunctional system of publishing and career advancement and thinking “how do I get everyone to sacrifice their freedom and sanity to propagate it?” instead of “how can I use my position and platform to get us out of this hellscape?”

10
6
77
56,151
Ben Hitz retweeted

17 May 2023
The @MonarchInit Biolink Model Brings Data Together! Find out more about our top 3 most downloaded papers in 2022 from the journal ASCPT here: ascpt.org/Journals/CTS/Trans…
#BiolinkModel #TeamScience #Collaboration #RareDisease #DrugRepurposing #TranslationalScience @linkml_data
4
2
465
Ben Hitz retweeted

17 May 2023
~Implement universal healthcare
~Untether health insurance from employment
~Invest in public health at point of need (meet people where they are)
~Adopt systems approach to elevate health literacy across all communities
(Sad typing this. Been saying same words for 10yrs.)
#hcldr
1
4
4
602
Ben Hitz retweeted

13 May 2023
Loved it.
It matters who does science science.org/content/blog-pos…
2
3
734
Ben Hitz retweeted

11 May 2023
If you publish papers your data (and code) has to be fully open for anyone to do as they please #OpenData #GreaterGood
(If you want to hide your data don't publish things 😉)
2
3
17
7,291