
4Dspacetime, sure,but it isn't the same as a 4Dspace. With this logic, you're saying we live in a higher dimensional world when this obviously isn't the case. So, in the context of higher dimensionality, it would matter here.I dont want to go back n fourth too much here we can dm
1
3
74

22 May 2025
Artificial Intelligence for Direct Prediction of Molecular Dynamics Across Chemical Space
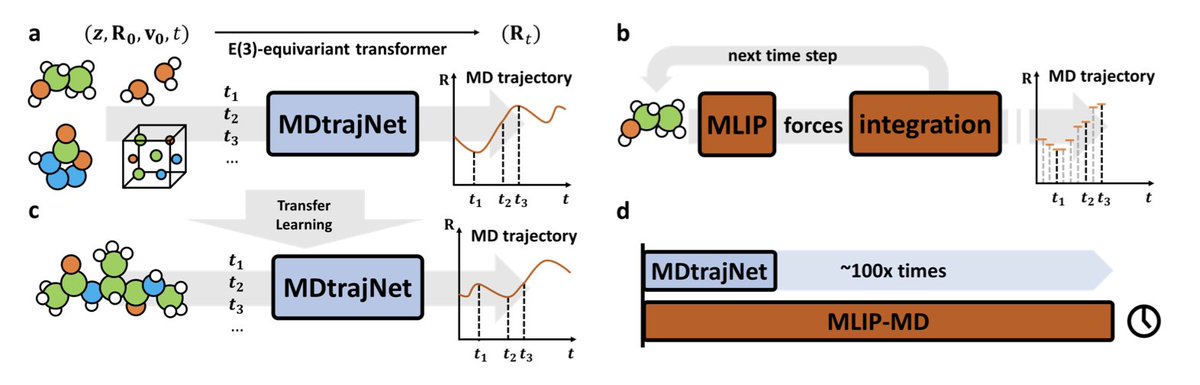
1.MDtrajNet-1 introduces a paradigm shift in molecular dynamics (MD) by directly predicting atomic trajectories in 4D spacetime—bypassing force calculations and time-step integration. This foundational model enables simulations that are over 100× faster than traditional MD, even those accelerated by ML force fields.
2.The model replaces iterative propagation with a Transformer-based, E(3)-equivariant neural network that learns to map atomic positions, velocities, element types, and time intervals directly to future geometries. This allows large-scale parallelization and efficiency without sacrificing accuracy.
3.MDtrajNet-1 achieves remarkable accuracy: in short-term predictions (10 fs), it reaches sub-picometer error levels, rivaling ab initio MD. For long-term predictions (10 ps), it reproduces spectral features with fidelity comparable to DFT-level simulations, despite being trained on a fraction of the data.
4.The model is trained on 1 million data points sampled from the ANI-1xMD dataset, covering 173 molecular systems with up to 9 atoms. Despite the limited size and diversity, MDtrajNet-1 generalizes well across unseen molecules and chemical spaces.
5.Compared to the GICnet model (an earlier proof-of-concept), MDtrajNet-1 demonstrates superior accuracy, scalability, and generalizability, thanks to architectural advances like multi-head equivariant attention and fine-grained representation of local atomic environments.
6.The model supports flexible ensemble conditions. Beyond NVE, MDtrajNet-1 can be retrained for NVT simulations and yields high-quality vibrational spectra even with added complexity from thermostats.
7.Its performance extends to periodic systems and different interaction types. It successfully simulates diamond lattices and Lennard-Jones fluids under periodic boundary conditions, capturing structural features like radial distribution functions.
8.MDtrajNet-1 also exhibits strong transfer learning capabilities. When fine-tuned on short trajectories from a 22-atom alanine dipeptide, it reproduces long-timescale conformational dynamics (Ramachandran plots) more accurately than MLIPs trained on the same data.
9.The model architecture is O(3)-equivariant, atom-centered, and scalable with system size. Computational time grows linearly with atom number, enabling efficient simulations of large systems on standard hardware (e.g., RTX 4090).
10.MDtrajNet-1 paves the way for general-purpose, foundational models in molecular simulation. Its blend of physics-inspired architecture and generative capability offers a compelling path toward scalable, accurate, and efficient trajectory generation in chemical and materials science.
💻Code: github.com/dralgroup/mlatom
📜Paper: doi.org/10.26434/chemrxiv-20…
#MolecularDynamics #4DSpacetime #TransformerModel #EquivariantNN #MDsimulation #AI4Science #ComputationalChemistry #MDtrajNet

3
17
1,150
22 May 2025
Artificial Intelligence for Direct Prediction of Molecular Dynamics Across Chemical Space
1.MDtrajNet-1 introduces a paradigm shift in molecular dynamics (MD) by directly predicting atomic trajectories in 4D spacetime—bypassing force calculations and time-step integration. This foundational model enables simulations that are over 100× faster than traditional MD, even those accelerated by ML force fields.
2.The model replaces iterative propagation with a Transformer-based, E(3)-equivariant neural network that learns to map atomic positions, velocities, element types, and time intervals directly to future geometries. This allows large-scale parallelization and efficiency without sacrificing accuracy.
3.MDtrajNet-1 achieves remarkable accuracy: in short-term predictions (10 fs), it reaches sub-picometer error levels, rivaling ab initio MD. For long-term predictions (10 ps), it reproduces spectral features with fidelity comparable to DFT-level simulations, despite being trained on a fraction of the data.
4.The model is trained on 1 million data points sampled from the ANI-1xMD dataset, covering 173 molecular systems with up to 9 atoms. Despite the limited size and diversity, MDtrajNet-1 generalizes well across unseen molecules and chemical spaces.
5.Compared to the GICnet model (an earlier proof-of-concept), MDtrajNet-1 demonstrates superior accuracy, scalability, and generalizability, thanks to architectural advances like multi-head equivariant attention and fine-grained representation of local atomic environments.
6.The model supports flexible ensemble conditions. Beyond NVE, MDtrajNet-1 can be retrained for NVT simulations and yields high-quality vibrational spectra even with added complexity from thermostats.
7.Its performance extends to periodic systems and different interaction types. It successfully simulates diamond lattices and Lennard-Jones fluids under periodic boundary conditions, capturing structural features like radial distribution functions.
8.MDtrajNet-1 also exhibits strong transfer learning capabilities. When fine-tuned on short trajectories from a 22-atom alanine dipeptide, it reproduces long-timescale conformational dynamics (Ramachandran plots) more accurately than MLIPs trained on the same data.
9.The model architecture is O(3)-equivariant, atom-centered, and scalable with system size. Computational time grows linearly with atom number, enabling efficient simulations of large systems on standard hardware (e.g., RTX 4090).
10.MDtrajNet-1 paves the way for general-purpose, foundational models in molecular simulation. Its blend of physics-inspired architecture and generative capability offers a compelling path toward scalable, accurate, and efficient trajectory generation in chemical and materials science.
💻Code: github.com/dralgroup/mlatom
📜Paper: doi.org/10.26434/chemrxiv-20…
#MolecularDynamics #4DSpacetime #TransformerModel #EquivariantNN #MDsimulation #AI4Science #ComputationalChemistry #MDtrajNet
1
12
991

31 Mar 2024
There is ample evidence extraterrestrials have been ,and are, visiting .Speed of light is not an issue bc they travel in a dimension outside of 4Dspacetime. U Tube "Preston Dennett" or Kindle " Crop Circles,Signs, Wonders & Mysteries" " for a start .
16

21 Jun 2019
You know about flat earth, but do you know about diamond-shaped-4Dspacetime Pac-Man earth?
3
11

10 May 2017
BENIGN: getting power from #crystals
SHINE: getting power from #metaphysical crystals, located outside of #4Dspacetime
1
19 Mar 2014
Your #clientside #experiencer/#perceiver life is more than just #4DSpaceTime; a reduced set of the even deeper realms of reality-at-large
1
1