
May 22
العقل البشري قد يساعد على فهم فرص التكامل بين النماذج اللغوية الضخمة ونماذج العالم كما يظهر هذا المشروع الشخصي لبناء روبوت صغير
youtube.com/watch?v=S67z2aek…
#WorldModel #LLM #TransformerModel #AI #Robotics #Trends
7
3,700

Mar 24
#Article: "Assessment of Non-Linear Modeling of Ladle Furnace Transformer Using Finite Element Analysis"
👨🏫 Author: Virna Costa Onofri, Thales Alexandre Carvalho Maia, Braz J. Cardoso Filho
👉 More information: mdpi.com/2075-1702/12/12/900
#FEA #TransformerModel

2
2
51

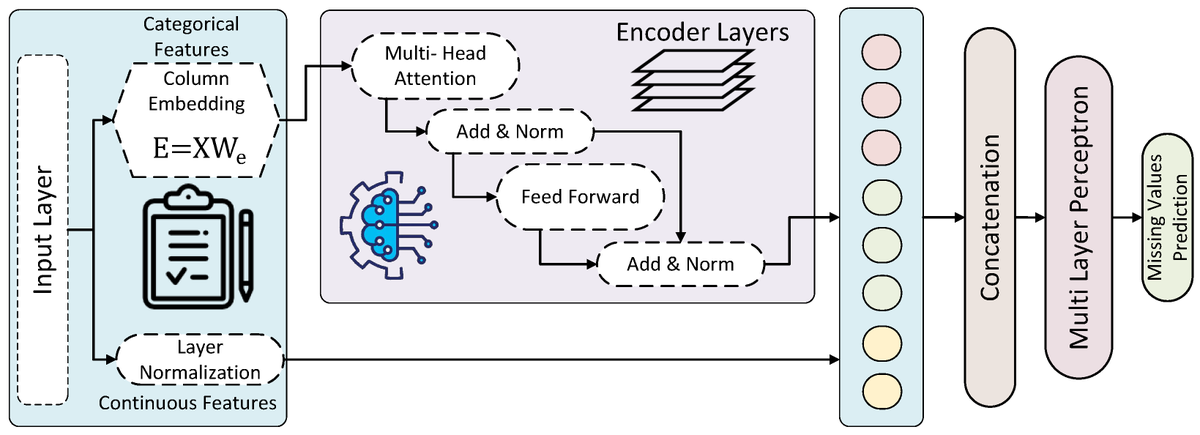
💥Highly recommended publication: "A Comparative Study on Imputation Techniques: Introducing a Transformer Model for Robust and Efficient Handling of Missing EEG Amplitude Data"
🔗shorturl.at/oUB2O
📌#TransformerModel #ClinicalData #DataQuality
4
38

My loss plateaued early in the training and I struggled fixing this the past 2 days. I finally solved it (thanks to Hugging Face Forums). I had to remove the dropout layers that I had sprinkled all over my layers and also decrease the learning rate
#attentionisallyouneed #transformermodel

2
7
989

22 Dec 2025
𝗠𝘆 𝗽𝗿𝗼𝗷𝗲𝗰𝘁 𝘀𝘂𝗽𝗲𝗿𝘃𝗶𝘀𝗼𝗿 𝘄𝗮𝘀 𝗯𝗲𝗻𝘁 𝗼𝗻 𝗼𝗻𝗲 𝘁𝗵𝗶𝗻𝗴.
Mr. Star would always say, “𝘠𝘰𝘶𝘳 𝘪𝘥𝘦𝘢 𝘥𝘪𝘥𝘯’𝘵 𝘥𝘳𝘰𝘱 𝘰𝘶𝘵 𝘰𝘧 𝘵𝘩𝘦 𝘴𝘬𝘺; 𝘴𝘰𝘮𝘦𝘰𝘯𝘦, 𝘴𝘰𝘮𝘦𝘸𝘩𝘦𝘳𝘦 𝘮𝘶𝘴𝘵 𝘩𝘢𝘷𝘦 𝘥𝘰𝘯𝘦 𝘴𝘰𝘮𝘦𝘵𝘩𝘪𝘯𝘨 𝘴𝘪𝘮𝘪𝘭𝘢𝘳.”
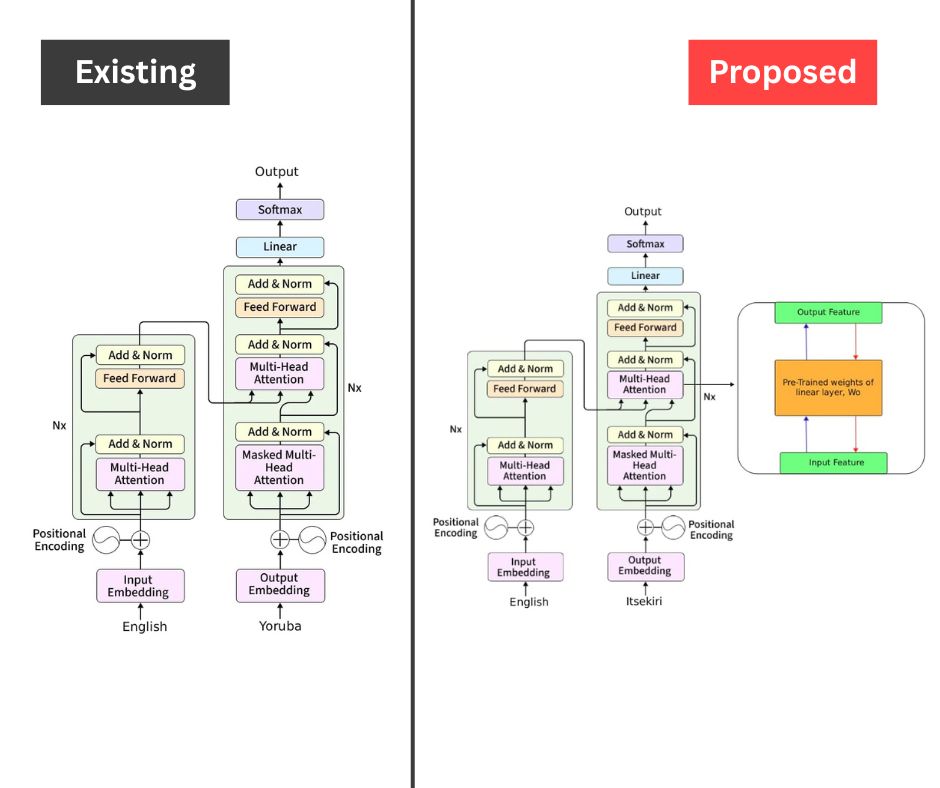
At first, this was quite confusing for me because after extensive research, I couldn’t find any paper written on a neural machine translator for English to Itsekiri. But my supervisor still kept insisting on identifying an existing system. He eventually suggested that while there may not be one for Itsekiri, there are many translators for other languages, so why not choose one of those as your reference system?
That’s when I realized that Itsekiri is a Yoruboid language, meaning it is a sub-group of the Yoruba language family. So I decided to use a system from a research paper written on an English-to-Yoruba translator by Mr Adeboje & Akinyede as my existing system.
Eventually, I went with the architecture shown in the picture. However, this architecture had a loophole: it was designed to work with large datasets. My major challenge during the course of the project was the limited availability of Itsekiri language data.
In the end, I resorted to fine-tuning, which is the process of adapting a large pre-trained model to perform well on a smaller, domain-specific dataset. This approach allows the model to retain its general language understanding while learning the nuances of the target language with limited data.
That’s why, in my proposed system, you’ll see the full fine-tuning feature integrated into the Transformer model architecture.
What do you guys think about my innovative architecture? 😁
#nlp #deeplearning #fineTuning #TransformerModel
3
40

12 Nov 2025
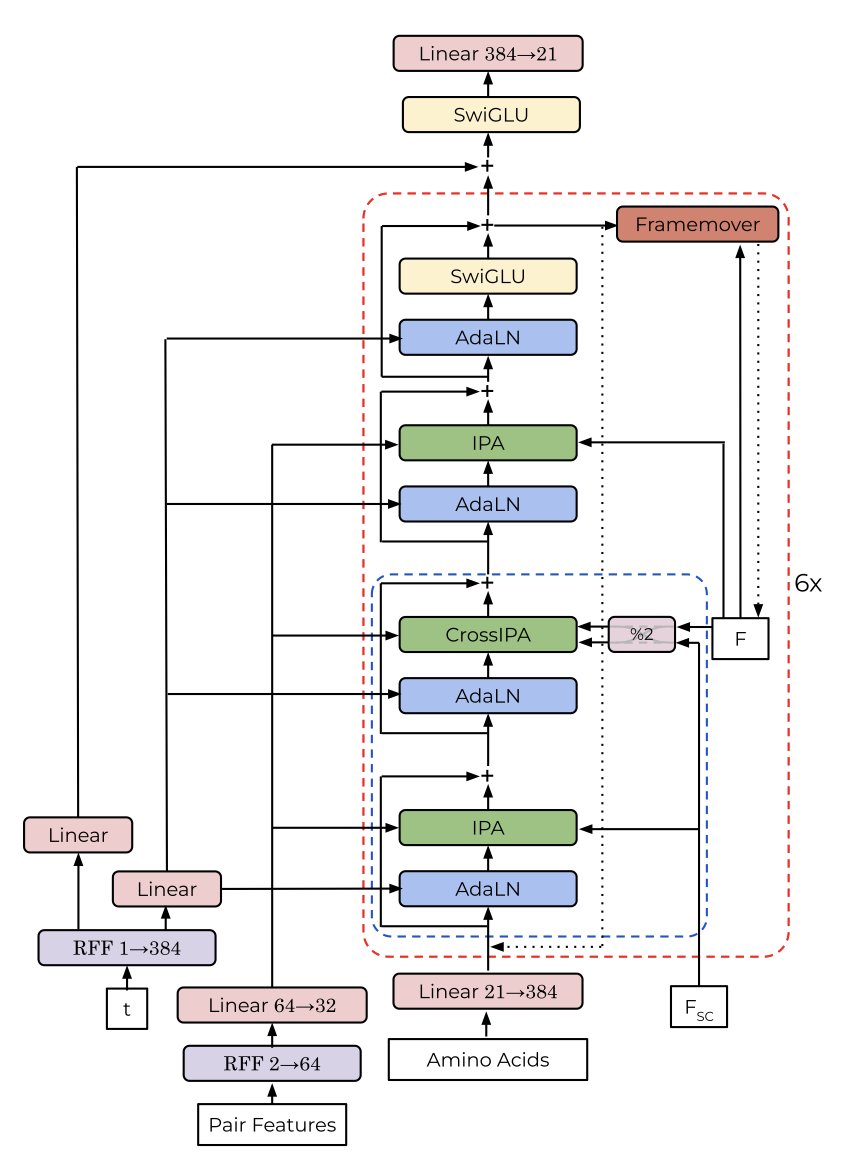
Spontaneous Emergence of Symmetry in a Generative Model of Protein Structure
1. A new study reveals that a transformer-based generative model can spontaneously generate symmetric protein structures without explicit symmetry constraints. This is a significant breakthrough in protein design as it demonstrates the model's ability to learn complex structural patterns implicitly from training data.
2. The model, called ChainStorm, uses flow matching and an SE(3)-equivariant transformer architecture. It was trained on multi-chain protein structures from the Protein Data Bank (PDB) without any symmetry-specific terms in the training objective. The emergence of symmetry was found to be driven by a single attention head in the model, which activates for pairs of residues that match across symmetric chains or repeating motifs within a chain.
3. The researchers discovered that the symmetry in generated structures is primarily determined early in the generative process and is influenced by matching chain lengths. When the lengths of the chains are similar, the model is more likely to generate symmetric structures. This finding suggests that the model can infer symmetry from basic structural information like chain length.
4. The study also demonstrates that suppressing the specific attention head responsible for symmetry prevents the model from generating symmetric structures, confirming its crucial role in symmetry emergence. This highlights the importance of understanding and controlling individual components of generative models to achieve desired design outcomes.
5. ChainStorm's ability to generate symmetric structures without explicit constraints opens new avenues for protein engineering. It suggests that future models could be designed to exploit such emergent behaviors for better control over protein design tasks, including the creation of multimers and self-assembling complexes.
6. The model was implemented in Julia and trained on a dataset of protein structures from the PDB. The training involved a novel approach to sampling and loss scaling, which helped stabilize the learning process and improve the model's performance in generating high-quality protein structures.
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/MurrellGroup/Chai…
#ProteinDesign #GenerativeModeling #Symmetry #TransformerModel #ComputationalBiology
1
3
8
1,124
20 Oct 2025
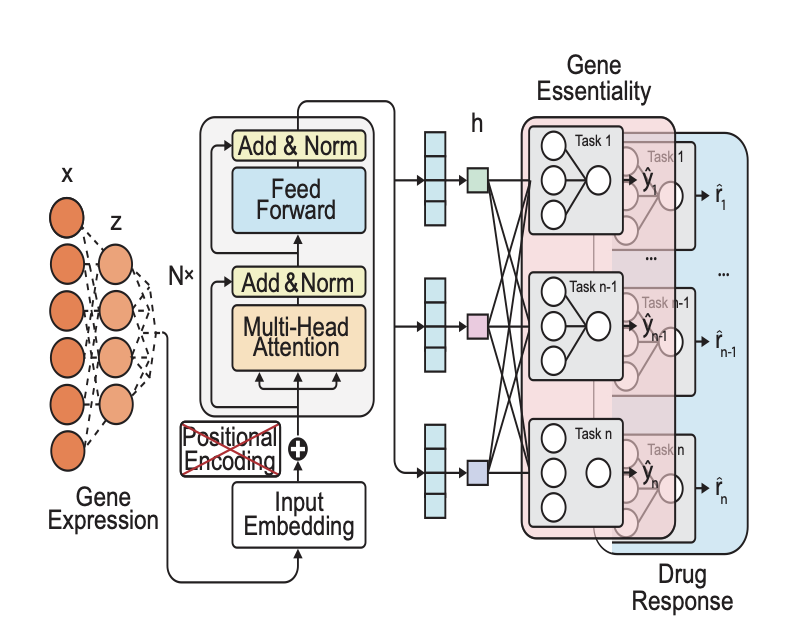
DeepVul: A Multi-Task Transformer Model for Joint Prediction of Gene Essentiality and Drug Response
1. DeepVul, a novel multi-task transformer-based model, has been proposed to predict gene essentiality and drug response from cancer transcriptome data, aiming to expand the applicability of precision oncology beyond the limited actionable genomic alterations.
2. The model aligns gene expressions, gene perturbations, and drug perturbations into a latent space, enabling simultaneous and accurate prediction of cancer cell vulnerabilities to numerous genes and drugs, which is a significant step forward in the field of precision medicine.
3. Benchmarking against existing precision oncology approaches revealed that DeepVul not only matches but also complements oncogene-defined precision methods, demonstrating its potential to enhance clinical actionability for a broader range of cancer patients.
4. Through interpretability analyses, DeepVul identifies underlying mechanisms of treatment response and resistance, as demonstrated with BRAF vulnerability prediction, providing valuable insights into the complex relationship between cancer genetic landscape and its vulnerabilities.
5. DeepVul’s ability to generalize predictions to new cohorts without additional training is a notable advantage, as shown by its performance on the Sanger Institute data, which was initially trained on DepMap essentiality data from the Broad Institute.
6. In predicting drug response via transfer learning, DeepVul tested two paradigms: one with a frozen expression encoder and another with a tunable encoder. Surprisingly, the model with the frozen encoder outperformed the tunable one, suggesting that gene essentiality might capture cancer at a more comprehensive systems level than drug response.
7. DeepVul was benchmarked against current actionable-mutation-based precision oncology approaches and demonstrated the ability to identify cell lines sensitive to targeting hundreds of genes with similar accuracy to existing precision oncology approaches, which is a major breakthrough in expanding the scope of precision oncology.
8. The interpretability framework of DeepVul could identify treatment response and resistance mechanisms by analyzing the correlation between SHAP values and gene expression levels, which may help in understanding the resistance mechanisms of targeted therapies.
💻Code: github.com/alaaj27/DeepVul.g…
📜Paper: biorxiv.org/content/10.1101/…
#DeepVul #PrecisionOncology #GeneEssentiality #DrugResponse #TransformerModel #ComputationalBiology #CancerTherapy
1
6
18
1,507
6 Oct 2025
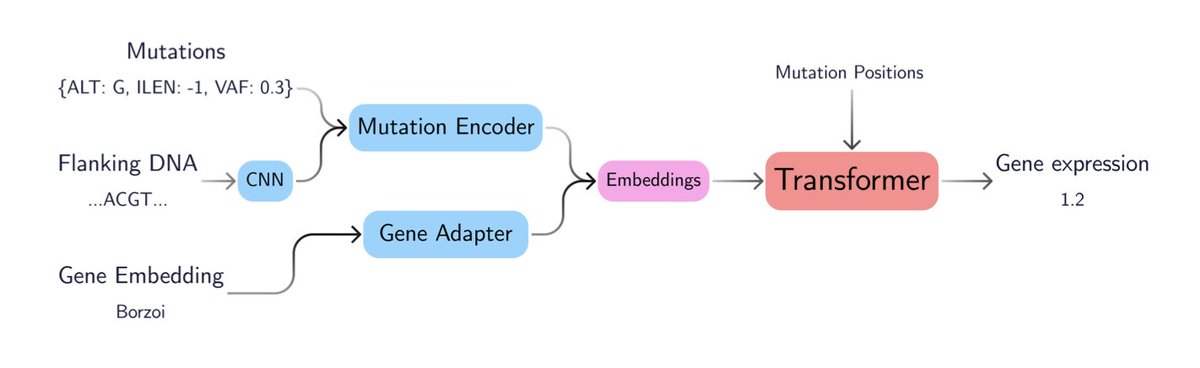
GenVarFormer: Predicting Gene Expression from Long-range Mutations in Cancer
1. GenVarFormer (GVF) is a novel transformer-based model that predicts gene expression from non-coding mutations in cancer, achieving a 26-fold higher correlation across samples than current models. This advancement significantly enhances the ability to identify driver mutations that fuel cancer progression.
2. GVF can efficiently model gene regulation across up to 16 million base pairs by focusing only on mutations and their local DNA context. This context window is 340 times larger than previous models, allowing it to capture sparse genetic variation and generalize to unseen genes and samples.
3. The model uses a novel approach to encode mutations, incorporating properties such as ALT, ILEN, VAF, flanking DNA, and POS. It employs rotary positional embeddings and a bin packing sampler to handle the extreme sparsity of somatic mutations and optimize GPU utilization.
4. GVF demonstrates superior performance in predicting gene expression, even in the most prevalent breast cancer subtype, luminal A. Patient embeddings generated by GVF are more informative for predicting overall patient survival than ground-truth gene expression data.
5. This work establishes a new state-of-the-art for modeling the functional impact of non-coding mutations in cancer. It provides a powerful tool for identifying potential driver events and improving patient prognosis. Future directions include incorporating germline variants and extending the model to other genetic diseases.
📜Paper: arxiv.org/abs/2509.25573v1
#GenVarFormer #CancerBiology #GeneExpression #NonCodingMutations #TransformerModel #Bioinformatics #CancerResearch
1
3
7
767
6 Oct 2025
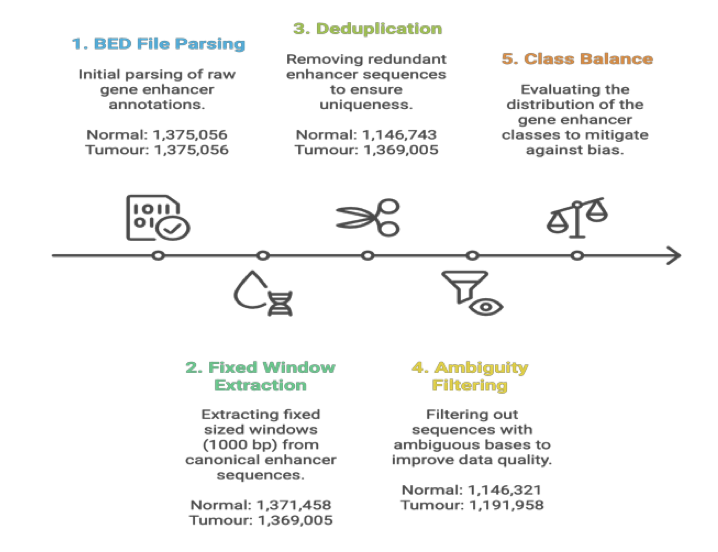
DNABERT-2: Fine-Tuning a Genomic Language Model for Colorectal Gene Enhancer Classification
1. A new study leverages DNABERT-2, a second-generation genomic language model, to classify colorectal gene enhancers with impressive results. This work demonstrates the potential of transformer-based models in capturing tumour-associated regulatory signals directly from DNA sequences alone, highlighting a novel path for cancer genomics research.
2. The study curates a balanced corpus of 2.34 million 1kb enhancer sequences from colorectal and normal tissue samples. Through meticulous preprocessing, including summit-centered extraction and rigorous deduplication, the dataset is well-prepared for training the DNABERT-2 model, ensuring high-quality input for the AI system.
3. DNABERT-2 employs byte-pair encoding (BPE) to tokenize DNA sequences, a significant innovation over previous fixed k-mer tokenization methods. This approach allows the model to capture both motif fragments and higher-order patterns more effectively, enhancing its ability to distinguish between normal and tumour-associated enhancers.
4. The model achieves a PR-AUC of 0.759, ROC-AUC of 0.743, and a best F1 score of 0.704 at an optimized threshold (τ = 0.359). These metrics indicate robust performance in ranking and identifying tumour enhancers, outperforming a CNN-based EnhancerNet in terms of recall and threshold-independent ranking.
5. The research underscores the importance of transformer architectures in genomic sequence analysis, showing that these models can move beyond motif-level encodings to provide holistic classification of regulatory elements. This advancement could lead to more accurate and comprehensive cancer genomics studies.
6. Future work will focus on improving precision, exploring hybrid CNN-transformer designs, and validating the model across independent datasets. These steps are crucial for strengthening the real-world applicability and generalizability of the DNABERT-2 model in cancer genomics.
📜Paper: arxiv.org/abs/2509.25274
#DNABERT2 #GenomicLanguageModel #ColorectalCancer #GeneEnhancerClassification #TransformerModel #CancerGenomics #ArtificialIntelligence
1
8
717
5 Oct 2025
ADAPT: Lightweight, Long-Range Machine Learning Force Fields Without Graphs
1. The article introduces ADAPT, a novel machine learning force field (MLFF) designed to address the limitations of existing graph neural network (GNN)-based models in capturing long-range interactions and avoiding oversmoothing when modeling point defects in materials.
2. ADAPT replaces graph representations with a direct coordinates-in-space formulation, treating atoms as "tokens" and using a Transformer encoder to model their interactions. This approach allows for explicit consideration of all pairwise atomic interactions, which is crucial for accurately predicting the properties of point defects.
3. The study demonstrates that ADAPT achieves a significant reduction in both force and energy prediction errors compared to state-of-the-art GNN-based models. Specifically, it shows a ∼33% reduction in errors on a dataset of silicon point defects, while requiring only a fraction of the computational resources.
4. ADAPT's architecture includes an importance-weighted mean squared error (MSE) loss function, which emphasizes the critical regions near defects during training. This specialized loss function helps improve the model's performance in practical applications where defect regions dominate the mechanical response of the material.
5. The training cost of ADAPT is remarkably lower than that of message-passing architectures. The small ADAPT model requires only 2.24 minutes per epoch on a single NVIDIA A100 GPU, converging after 80 epochs, whereas retraining MACE requires 8.5 minutes per epoch for 300 epochs on 16 NVIDIA A100 GPUs.
6. The authors also developed a separate formation energy-predictor model using a MLP residual architecture, which outperformed both MACE and MatterSim in predicting defect formation energies. This model achieves a better than 30% reduction in mean absolute error (MAE) over MatterSim 5M after 400 epochs of training.
7. ADAPT's design allows for independent deployment of the force and energy prediction models when only one quantity is needed, reducing runtime and memory consumption. This modularity also enables incremental model refinements without retraining the entire model.
8. The article highlights that while ADAPT's separation of force and energy predictions offers practical advantages, it results in a non-conservative MLFF where forces are not guaranteed to correspond to gradients of the energy surface. This trade-off is important to consider for applications requiring conservative force fields.
9. The authors suggest future directions for ADAPT, including enforcing physical invariances within the architecture and loss function, extending training to a wider class of defects and materials, and developing models that integrate physical constraints directly into the architecture.
📜Paper: arxiv.org/abs/2509.24115
#MachineLearning #MaterialsScience #PointDefects #TransformerModel #ComputationalEfficiency #ForceFields

1
2
11
1,332
5 Oct 2025
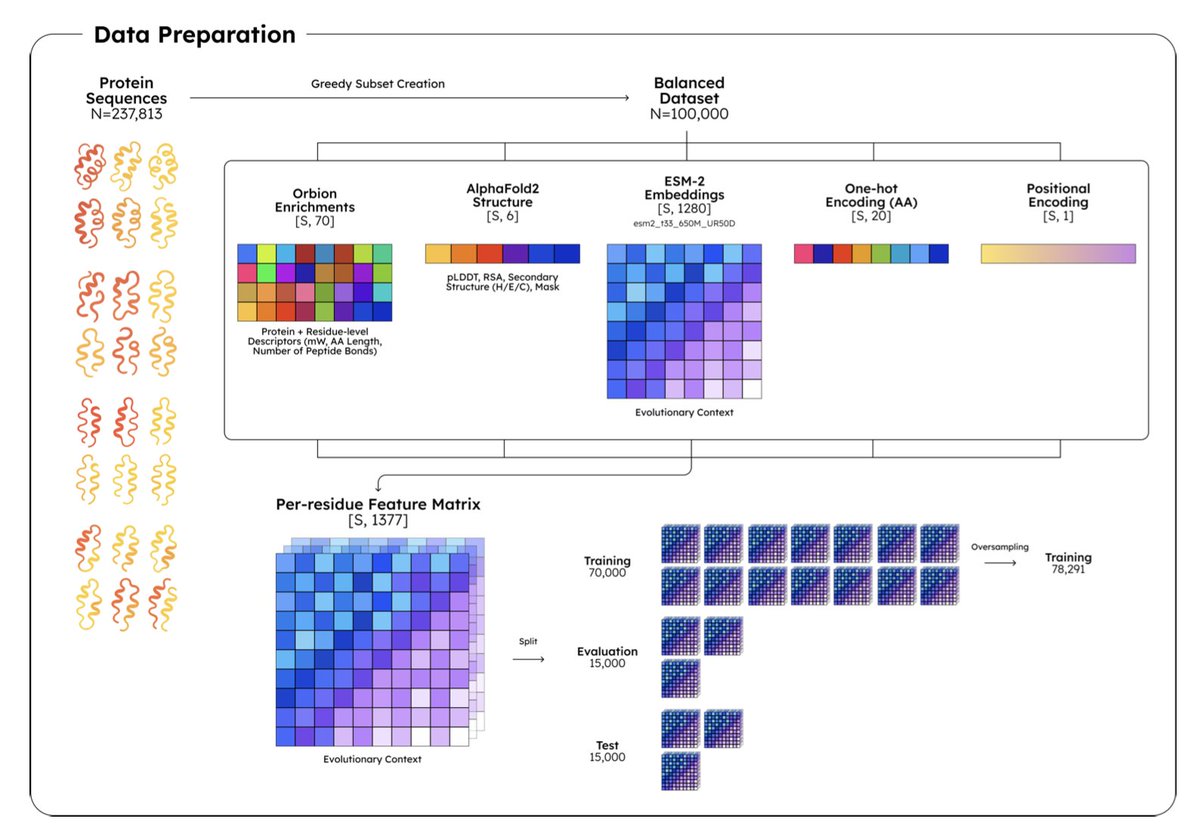
AstraPTM2: A Context-Aware Transformer for Broad-Spectrum PTM Prediction
1. AstraPTM2 is a novel computational tool that predicts 39 distinct types of post-translational modifications (PTMs) on full-length protein sequences. This broad coverage is unprecedented and addresses a significant gap in the field, as most existing tools focus on a limited number of PTMs or truncate protein sequences, losing critical context.
2. The model integrates ESM-2 embeddings, AlphaFold2-derived structural features, and protein-level descriptors to capture both short-range motifs and long-range dependencies. This multi-scale approach allows AstraPTM2 to identify PTMs that are driven by complex structural and sequence contexts, enhancing its predictive power.
3. AstraPTM2 employs a three-stage curriculum learning strategy to balance the prediction of rare and common PTMs. By progressively introducing rarer labels during training, the model achieves a macro-F1 score of 59% and an AUROC of 0.99, demonstrating strong performance across a diverse set of PTMs, including those with limited training data.
4. The model’s outputs are calibrated using per-label affine calibration and optimized thresholds, ensuring that predictions are reliable and interpretable. This calibration process is crucial for generating actionable insights, especially for experimental biologists planning mutagenesis studies or designing protein constructs.
5. AstraPTM2 is deployed on the Orbion web platform, offering synchronized 2D and 3D visualizations of PTM predictions. The platform supports dual prediction modes—calibrated and exploratory—allowing users to distinguish high-confidence sites from lower-confidence hypotheses, facilitating both decision-making and hypothesis generation.
6. In hold-out tests, AstraPTM2 shows particularly strong performance on rare motif-driven PTMs such as O-linked glycosylation and sumoylation. This highlights the model’s ability to identify PTMs that are often overlooked by other tools, potentially revealing new biological insights and therapeutic targets.
7. The development of AstraPTM2 underscores the importance of balanced data curation, thoughtful curriculum scheduling, and motif-sensitive architectures. These principles are likely to guide the next generation of multi-PTM predictors, emphasizing that thoughtful design can be more impactful than simply increasing model size or dataset volume.
📜Paper: biorxiv.org/content/10.1101/…
#AstraPTM2 #PTMPrediction #ComputationalBiology #TransformerModel #ProteinStructure #MachineLearning #Bioinformatics
1
3
14
1,599
4 Sep 2025
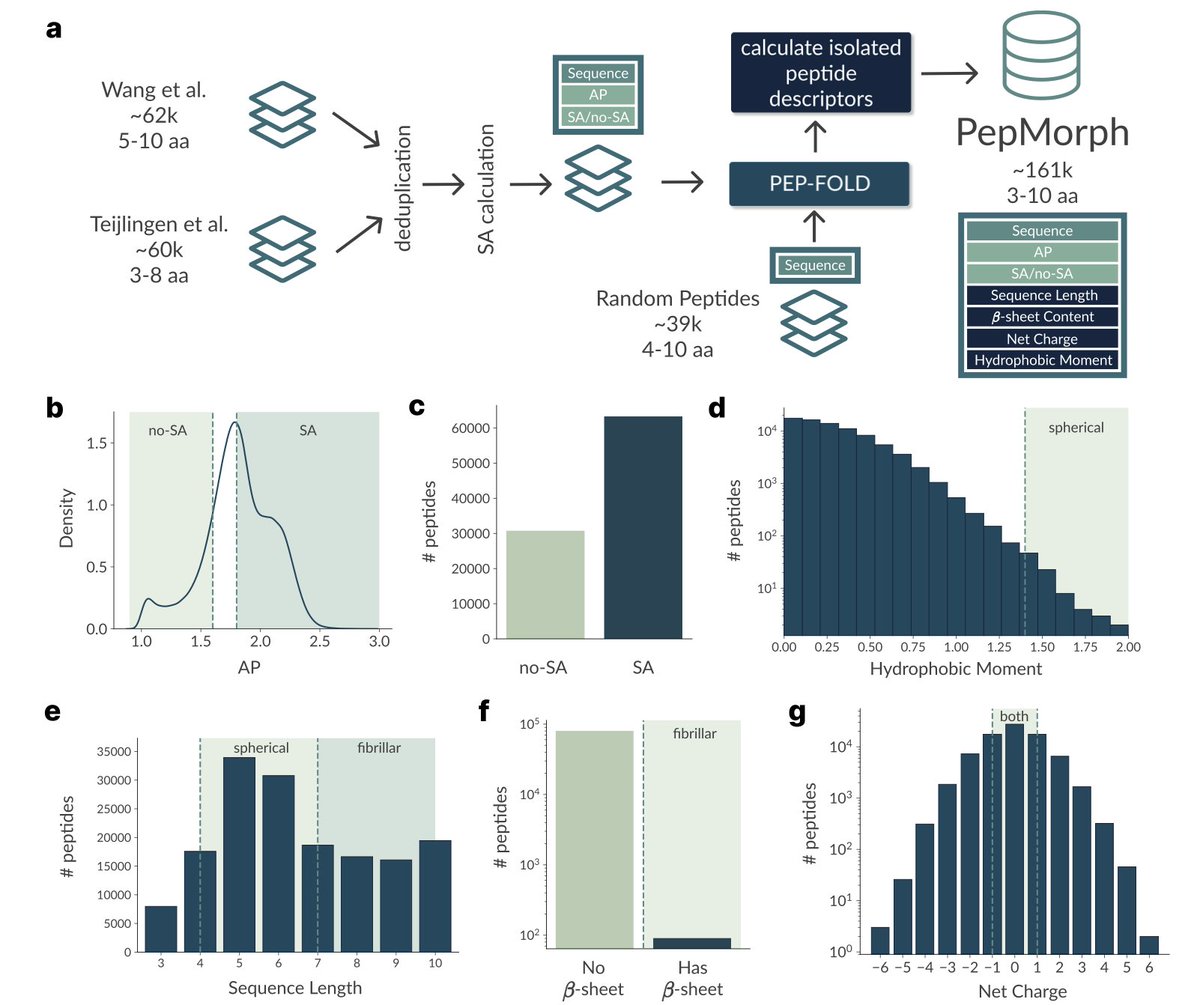
Morphology-Specific Peptide Discovery via Masked Conditional Generative Modeling
1. A novel study introduces PepMorph, an end-to-end pipeline for discovering peptides that self-assemble into specific morphologies like fibers or spheres. This is a significant step forward in designing biocompatible materials with tailored properties for biomedical and energy applications.
2. PepMorph leverages a Transformer-based Conditional Variational Autoencoder with a masking mechanism. This allows the model to generate novel peptides under arbitrary conditioning, making it highly flexible for targeting different morphologies based on geometric and physicochemical descriptors.
3. The study compiles a new dataset by integrating existing aggregation propensity datasets and extracting peptide descriptors that act as proxies for aggregate morphology. This dataset is crucial for training the model and ensuring it can generate peptides with desired properties.
4. PepMorph demonstrates high novelty and diversity in generated sequences, with less than 10% similarity among generated peptides. The model achieves 83% accuracy in intended morphology generation, validated through coarse-grained molecular dynamics simulations.
5. The framework is highly versatile and can be extended to longer peptides and more complex morphologies. It also allows for the integration of additional descriptors, making it a powerful tool for future peptide design and discovery efforts.
6. The study highlights the importance of the masking mechanism in handling partial conditioning, which is essential for generating peptides that meet specific design criteria without collapsing to a few templates.
7. The authors provide the curated PepMorph dataset and simulation trajectories publicly, along with the trained model weights and code, facilitating further research and development in this exciting field.
💻Code: github.com/tummfm/pepmorph
📜Paper: arxiv.org/abs/2509.02060v1
#PeptideDiscovery #MolecularDesign #GenerativeModeling #BiocompatibleMaterials #MolecularDynamics #TransformerModel #MachineLearning #ComputationalBiology
1
2
11
1,511
26 Aug 2025
HemePLM–Diffuse: A Scalable Generative Framework for Protein–Ligand Dynamics in Large Biomolecular System
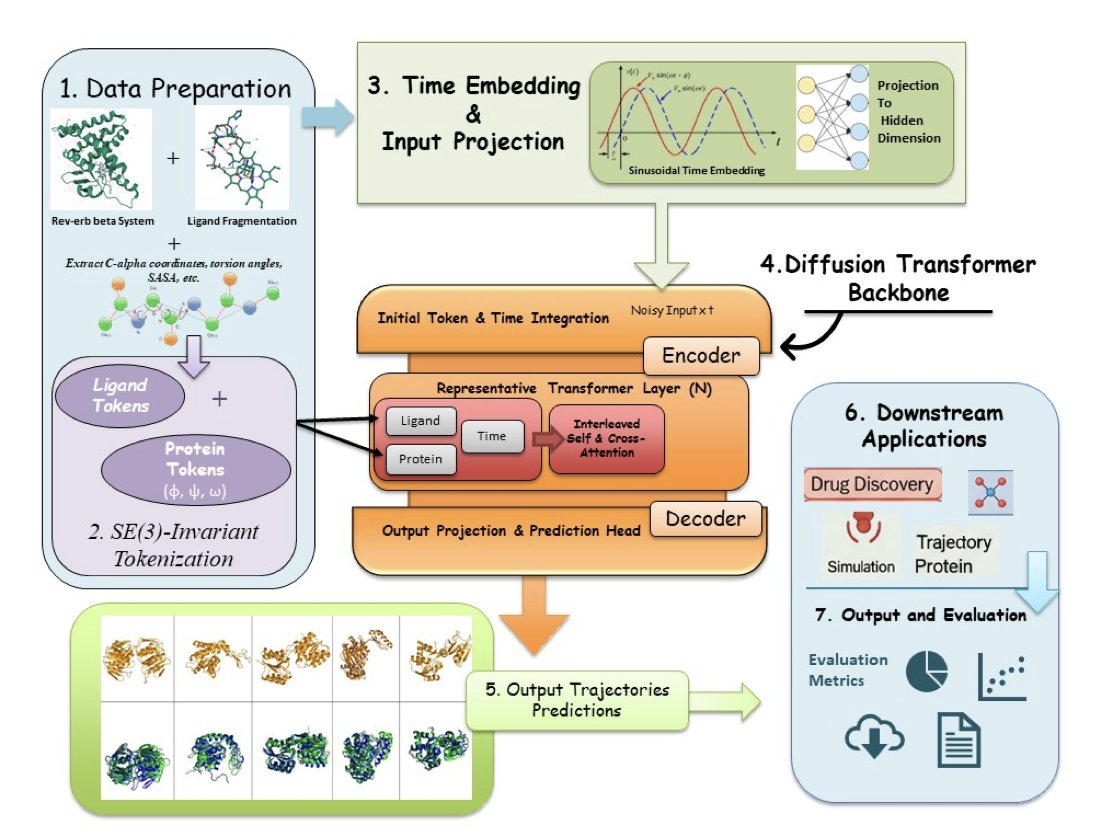
1. HemePLM-Diffuse is a novel generative transformer model designed to simulate the long-timescale dynamics of protein–ligand complexes in systems with over 10,000 atoms, addressing a significant challenge in computational biology and drug discovery.
2. The model introduces an SE(3)-invariant tokenization approach for both proteins and ligands, ensuring accurate representation of 3D molecular geometry. This is combined with a time-aware cross-attentional diffusion mechanism to effectively capture atomic motion over extensive spatiotemporal scales.
3. HemePLM-Diffuse demonstrates superior performance in ligand inpainting, trajectory upsampling, and transition path sampling compared to leading models like TorchMD-Net, MDGEN, and Uni-Mol. It achieves an average RMSD of 0.91 Å in ligand inpainting, significantly lower than previous models.
4. The model’s trajectory upsampling capability allows for the faithful recovery of atomic dynamics between known states, with an average RMSD of 1.03 Å per frame. This is crucial for computationally expensive systems where frequent MD snapshots are impractical.
5. In transition path sampling, HemePLM-Diffuse generates plausible intermediate frames that preserve contact maps and ligand orientation, achieving a TPS score of 0.95. This highlights its ability to simulate realistic conformational switches.
6. HemePLM-Diffuse achieves a speedup of over 100× compared to traditional MD simulations, with a simulation runtime of approximately 12 minutes for 1 ns equivalent dynamics on a large system like 3CQV HEME. This efficiency is enabled by parallelized transformer rollouts and the absence of integration steps.
7. Despite its advancements, the model currently does not simulate solvent molecules explicitly, which is a limitation for more complex environments. Future work includes extending the model to multi-ligand systems, ensemble-based fine-tuning, and integration with cryo-EM/NMR data.
📜Paper: arxiv.org/abs/2508.16587v1
#ComputationalBiology #DrugDiscovery #ProteinLigandDynamics #GenerativeModeling #TransformerModel #MolecularDynamics
1
2
791
23 Aug 2025
xBitterT5: An Explainable Transformer-Based Framework with Multimodal Inputs for Identifying Bitter-Taste Peptides @jcheminf
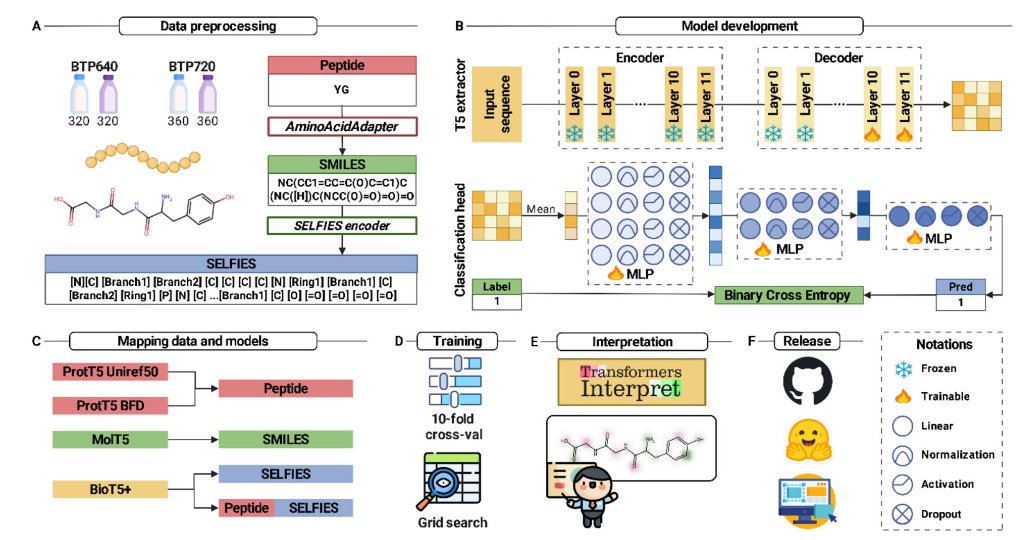
1. Nguyen et al. have introduced xBitterT5, a novel framework for identifying bitter peptides (BPs) using a combination of peptide sequences and their molecular representations. This multimodal approach leverages the pretrained BioT5 model to integrate peptide sequences with their SELFIES molecular representation, offering superior performance and interpretability compared to previous methods.
2. The study highlights the importance of bitter peptides in both food science and biomedicine. BPs play a crucial role in taste perception and various physiological processes. Accurate identification of these peptides is essential for understanding food quality and potential health impacts. xBitterT5 addresses the limitations of traditional machine learning approaches by providing a more comprehensive understanding of peptide sequences through their molecular strings.
3. A key innovation of xBitterT5 is its ability to provide residue-level interpretability. The model can identify specific molecular substructures within a peptide that contribute to its bitterness, offering valuable insights into the structure–activity relationship of BPs. This interpretability is crucial for peptide-based food and drug applications, as it goes beyond mere predictions to provide mechanistic insights.
4. The researchers conducted comprehensive evaluations on two benchmark datasets, BTP640 and BTP720. xBitterT5 demonstrated significant improvements in performance metrics such as accuracy, specificity, precision, sensitivity, F1 score, and the area under the receiver operating characteristic (ROC) curve. The model’s ability to generalize well to unseen data was also validated through independent testing.
5. To facilitate accessibility and reproducibility, the authors have developed a user-friendly web server and made the source code and trained models publicly available. This enables both computational biologists and experimental researchers to utilize xBitterT5 for BP identification, fostering further advancements in peptide-based research.
6. The study concludes that xBitterT5 not only advances the field of BP identification but also serves as a foundation for broader applications in peptide-centric research. Future work could involve expanding the model’s scope to other flavor-associated peptides and exploring alternative molecular string representations to enhance its capabilities further.
📜Paper: jcheminf.biomedcentral.com/a…
💻Code: github.com/cbbl-skku-org/xBi…
#xBitterT5 #BitterPeptides #MultimodalInputs #TransformerModel #PeptideIdentification #Bioinformatics #MachineLearning #ComputationalBiology
1
3
10
1,478
16 Aug 2025
Designing Lipid Nanoparticles Using a Transformer-Based Neural Network @NatureComms
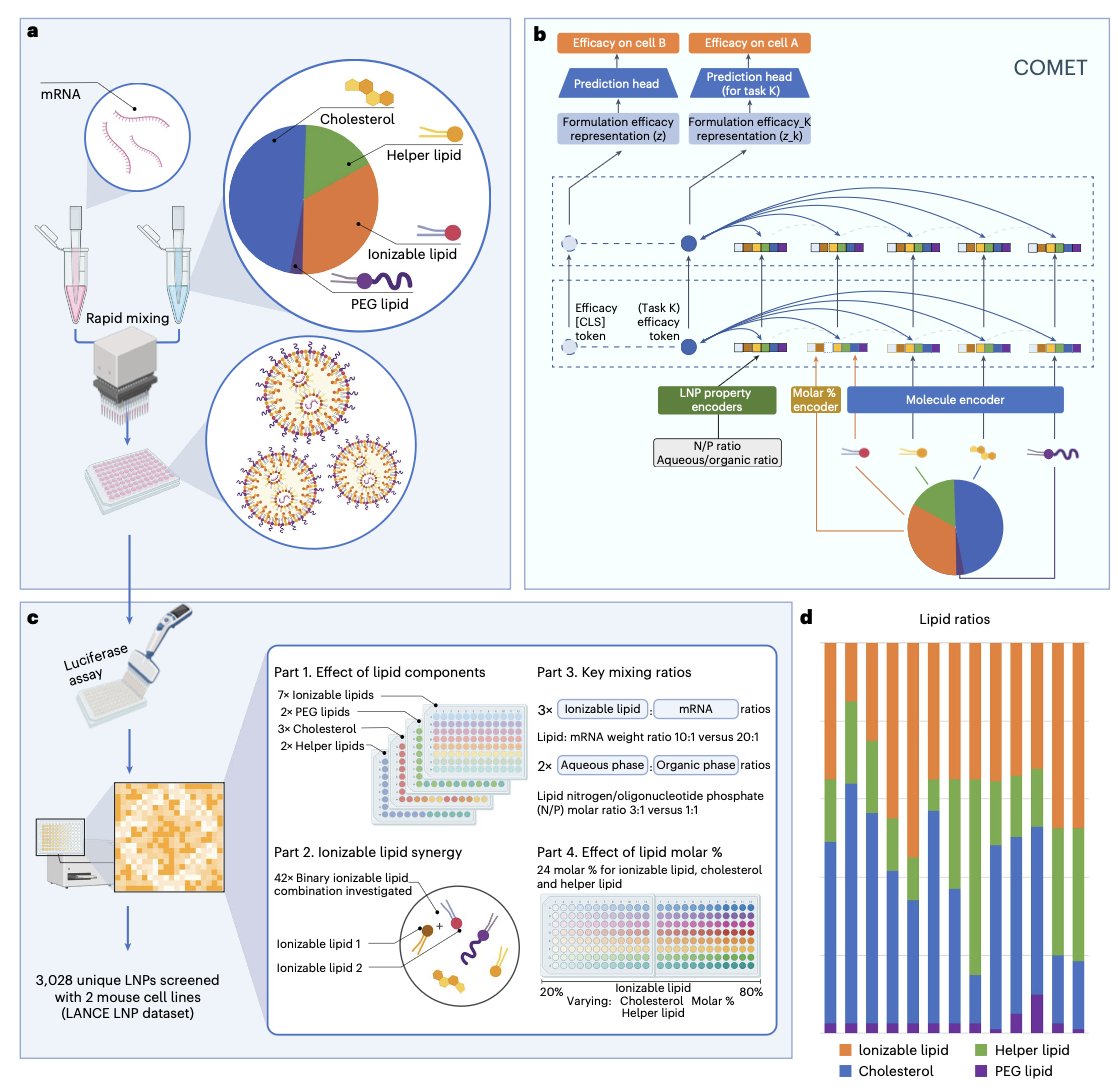
1. Researchers have developed a novel approach to designing lipid nanoparticles (LNPs) using a transformer-based neural network named COMET. This model integrates multi-component and multi-modal features of composite formulations to predict LNP performance accurately.
2. COMET is trained on one of the largest LNP datasets, LANCE, which includes over 3,000 unique LNPs. The model not only predicts efficacy but also adapts to non-canonical LNP formulations, such as those with dual-ionizable lipids and polymeric materials.
3. A key innovation is COMET's ability to predict LNP performance in new cell lines and under different conditions, such as post-lyophilization stability, using only small training datasets. This adaptability makes it a versatile tool for drug development.
4. Experimental validation shows that COMET can identify LNPs with strong protein expression both in vitro and in vivo, demonstrating its potential to accelerate the development of nucleic acid therapies across various applications.
5. The study highlights the importance of integrating molecular structures, molar percentages, and synthesis parameters into a unified framework. COMET's architecture allows for data-driven learning without relying on manually selected physicochemical descriptors.
6. COMET's flexibility extends to exploring non-canonical formulations and optimizing existing leads. It can screen massive virtual libraries to find formulations that differ substantially from known hits yet yield high performance.
7. The model's predictions are validated through in vitro and in vivo experiments, showing significant improvements over clinical benchmarks. COMET's ability to handle multi-component inputs also allows for its extension beyond conventional LNPs to other areas of nanotechnology.
📜Paper: nature.com/articles/s41565-0…
#LipidNanoparticles #TransformerModel #DrugDevelopment #MachineLearning #NucleicAcidTherapies
5
1,212
10 Aug 2025
MoCETSE: A Mixture-of-Convolutional Experts and Transformer-Based Model for Predicting Gram-Negative Bacterial Secreted Effectors
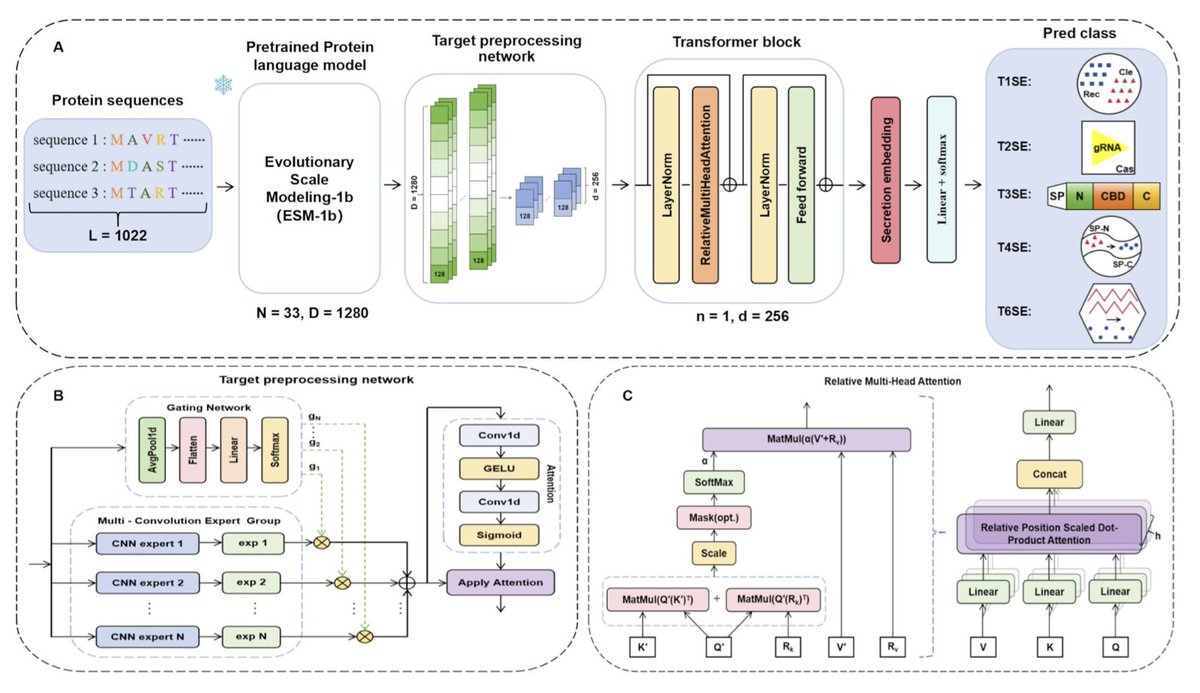
1. MoCETSE is a novel deep learning model designed to predict effector proteins from Gram-negative bacterial secretion systems with high accuracy. This model leverages a combination of convolutional neural networks and transformers to capture both local and long-range dependencies in protein sequences.
2. The model incorporates a pre-trained protein language model (ESM-1b) to transform raw amino acid sequences into context-aware vector representations. This step is crucial for extracting meaningful biological features from the sequences.
3. A key innovation in MoCETSE is the use of a target preprocessing network based on a mixture of convolutional experts. This architecture allows parallel processing of data by multiple convolutional kernels, which helps in learning diverse local motifs and broader contextual information.
4. MoCETSE integrates relative positional encoding into its transformer module. This feature enhances the model’s ability to recognize sequential relationships and long-range functional dependencies among amino acids, improving prediction accuracy.
5. The model demonstrates superior performance in both 5-fold cross-validation and independent testing, outperforming existing methods such as DeepSecE. It achieves high AUC and AUPRC values, indicating excellent discrimination between effector and non-effector proteins.
6. MoCETSE also provides biological interpretability by identifying key sequence motifs related to secretion signals. This is achieved through the relative position multi-head attention mechanism, which highlights important regions in the protein sequences.
7. The study highlights the potential of MoCETSE to be used as a high-throughput method for effector protein prediction, aiding in the understanding of bacterial pathogenic mechanisms and the development of antimicrobial strategies.
📜Paper: biorxiv.org/content/10.1101/…
#DeepLearning #ProteinPrediction #BacterialEffectors #Bioinformatics #TransformerModel #ConvolutionalNeuralNetworks
1
1
7
1,136
4 Aug 2025
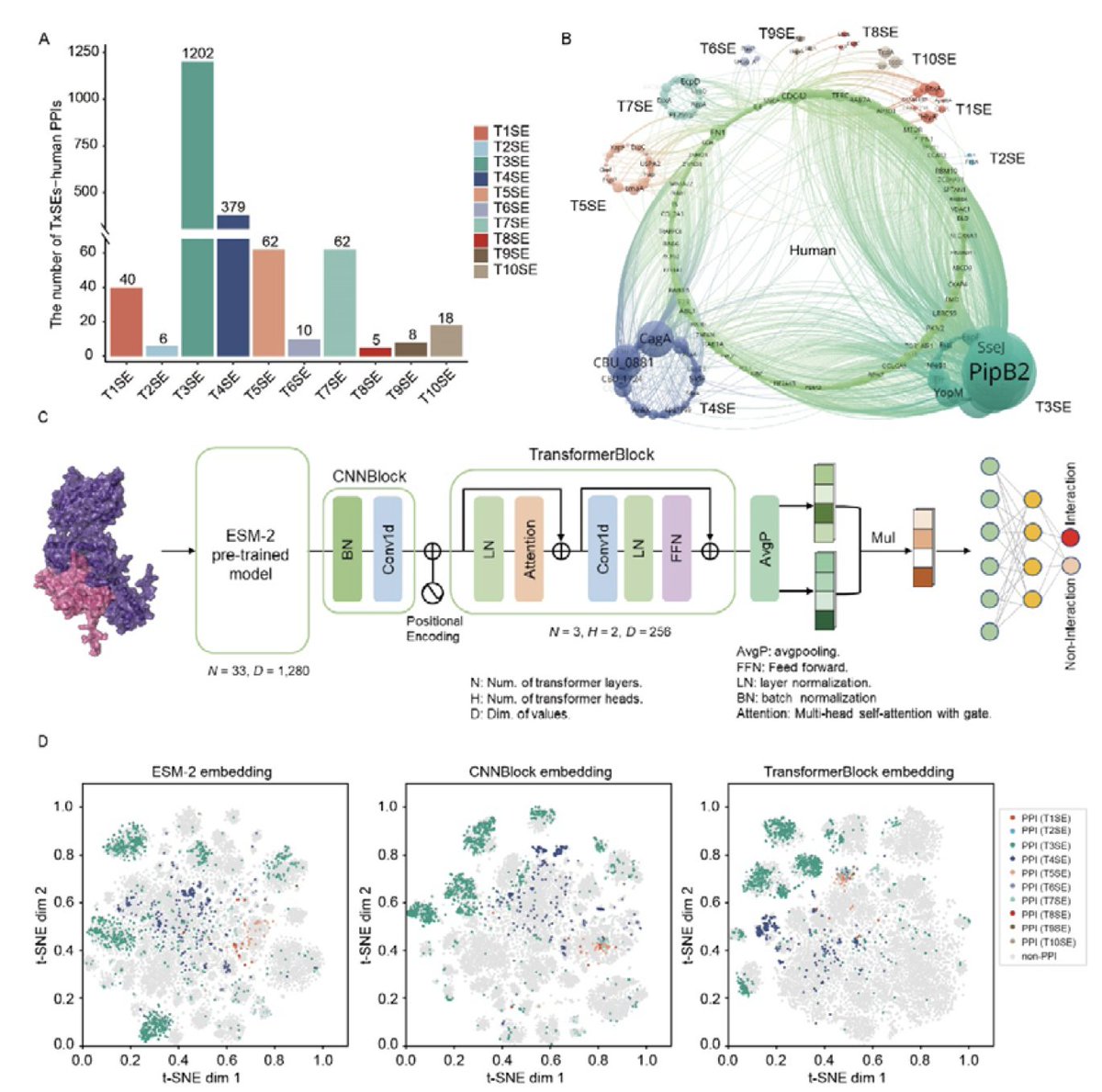
HPInet: Interpretable prediction of Host-Pathogen protein-protein Interactions using a transformer-based neural network
1. HPInet is a novel transformer-based deep learning model designed to predict interactions between bacterial effector proteins and human host proteins, achieving significant improvements in accuracy and sensitivity compared to existing methods.
2. The model integrates convolutional layers and global response normalization into its architecture, enhancing the extraction of both local and global features from protein sequences. This innovative approach allows HPInet to capture complex interaction patterns more effectively.
3. HPInet demonstrates superior performance across various bacterial secretion systems, with notable improvements in cross-system predictions, particularly between T5SE and T7SE. This highlights its ability to generalize while retaining system-specific features.
4. One of the key strengths of HPInet is its interpretability. By leveraging attention mechanisms, the model identifies critical interaction residues, providing valuable biological insights that align with experimentally validated binding sites.
5. HPInet is freely accessible as a web server, offering researchers a powerful and user-friendly tool to study host-pathogen interactions and identify potential therapeutic targets. This platform is expected to significantly advance research in bacterial pathogenesis.
📜Paper: biorxiv.org/content/10.1101/…
#HostPathogenInteraction #TransformerModel #DeepLearning #ProteinInteraction #Bioinformatics #ComputationalBiology
10
28
1,577

2 Aug 2025
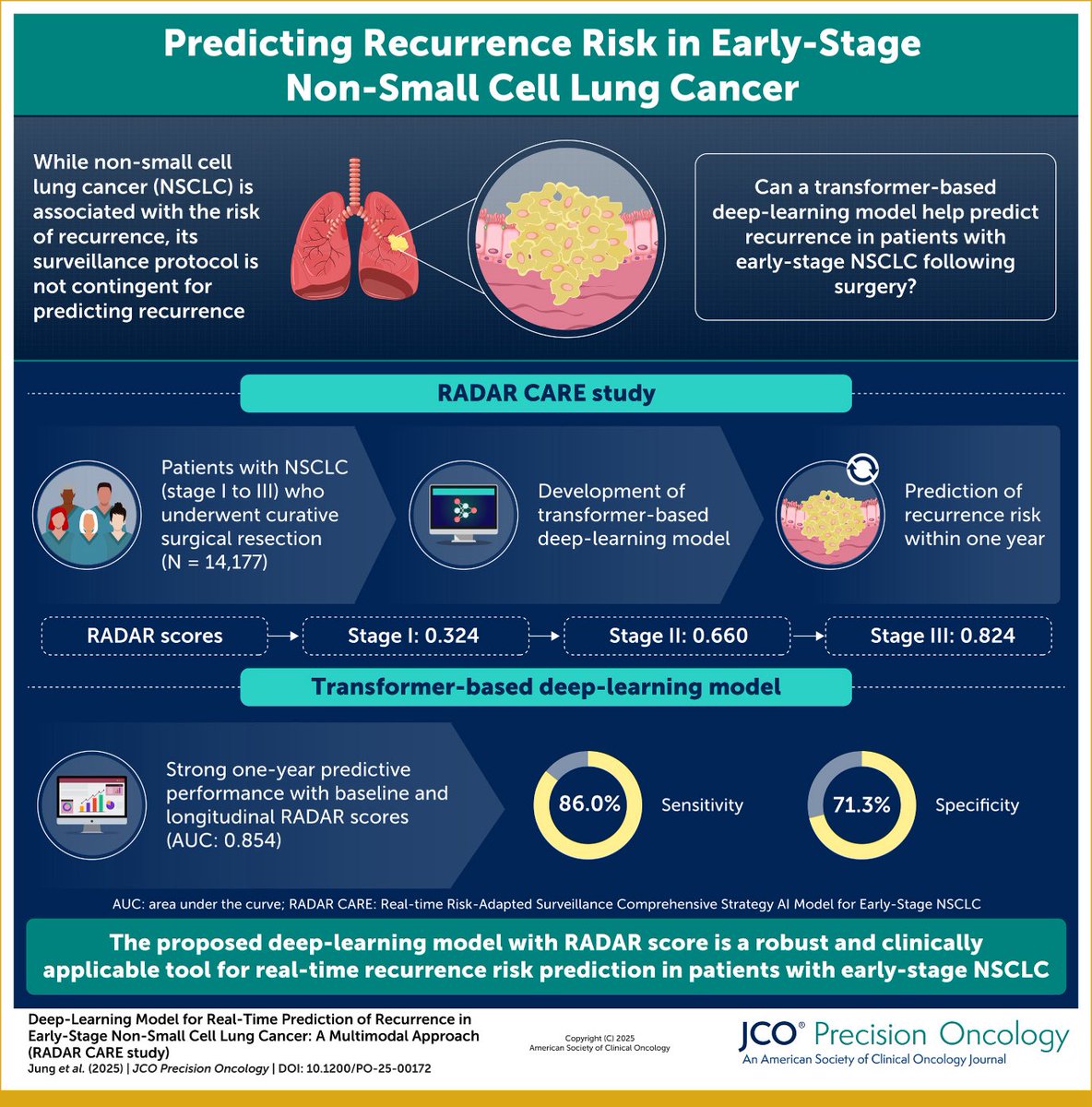
Can AI predict cancer recurrence before it happens?
📢 A new deep-learning model (RADAR CARE) predicts 1-year recurrence after curative surgery in early-stage NSCLC using multimodal clinical & radiologic data.
✅ 14,177 patients
✅ AUC: 0.854
✅ Sensitivity: 86%, Specificity: 71.3%
✅ Risk score independent of TNM stage
🧠 RADAR model uses transformer-based AI to process 64 clinical, molecular & imaging features, providing real-time recurrence risk with actionable thresholds:
•Low risk: RADAR < 0.3 (1-year recurrence <1%)
•Intermediate: 0.3–0.6
•High risk: >0.6 (recurrence ≥5%)
🔁 Four dynamic risk patterns suggest how to tailor follow-up & adjuvant therapy.
📌 Implication: Surveillance in early-stage NSCLC can now be personalized, moving beyond rigid TNM-based protocols.
📎 ascopubs.org/doi/full/10.120…
#LungCancer #NSCLC #Oncology #AIinHealthcare
#PrecisionOncology #DeepLearning #RecurrenceRisk
#CancerSurveillance #ThoracicOncology #RadOnc
#JCOPO #CancerAI #TransformerModel #MedicalAI
@JCO_ASCO @JCOGO_ASCO @ASCO @crisbergerot @osutcuoglu @Erman_Akkus @BbaharK @RyanNipp @brunolarvol @OncoAlert @oncodaily @OncoReporte @realbowtiedoc @ErulEnes @Xesiloglu @kbaskurtt
1
10
26
4,860

1 Aug 2025
I thought a bit about the spam detection algorithm on X's side and the characteristics of Grok, but I think there's probably nothing we can do about this unless we wait for the Attention to be reset on X's side. I mainly tinker with GPT systems, but in the end it's a TransformerModel so I kind of get it.
1
2
789
4 Jul 2025
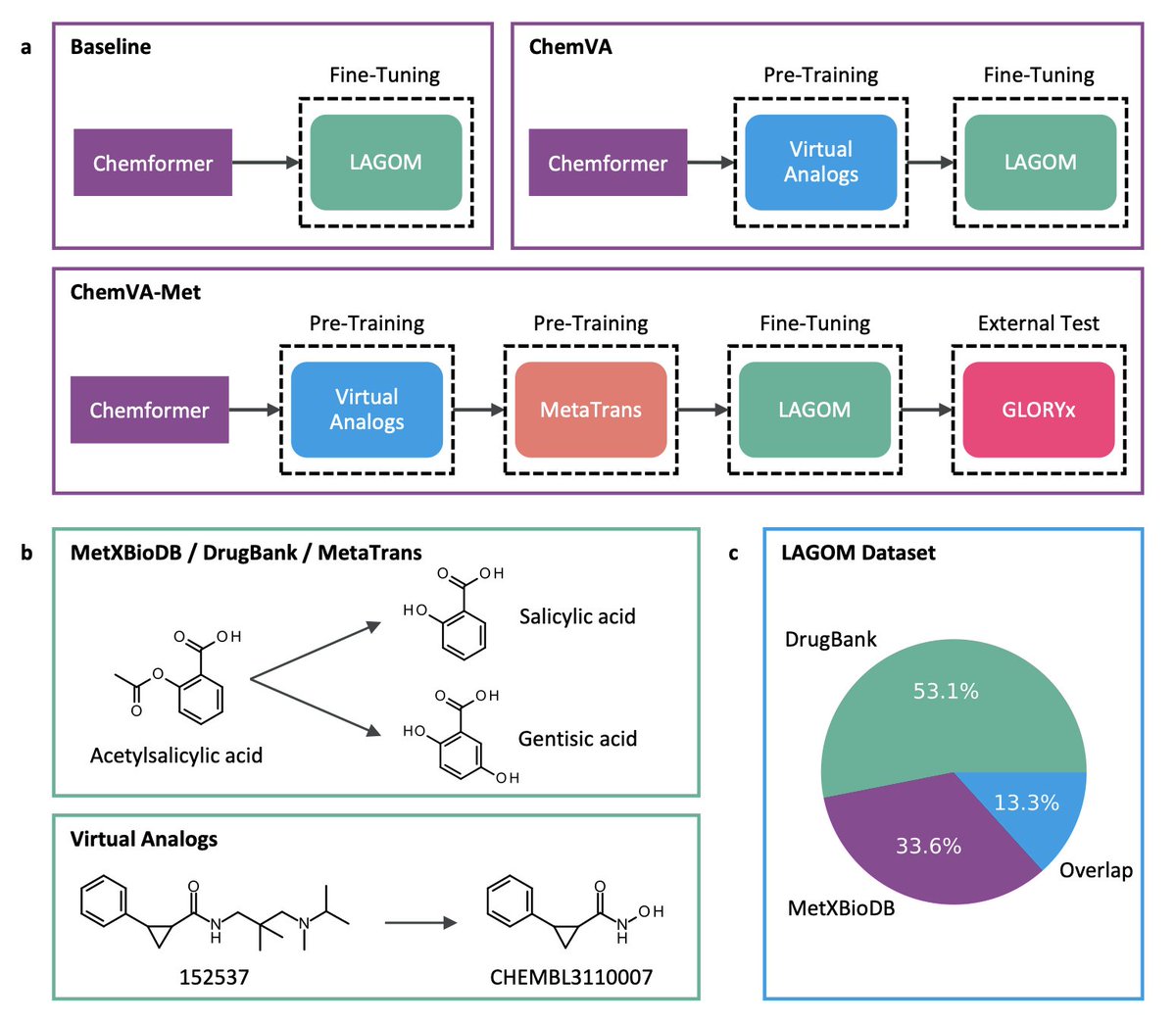
LAGOM: A Transformer-Based Chemical Language Model for Drug Metabolite Prediction
1.LAGOM is a Transformer-based model designed to predict drug metabolites directly from SMILES, offering an end-to-end alternative to traditional rule-based and two-step ML approaches.
2.Unlike conventional tools that rely heavily on manually curated transformation rules and intermediate site-of-metabolism (SoM) predictions, LAGOM leverages the Chemformer architecture to perform direct sequence-to-sequence translation from drugs to metabolites.
3.The model is trained using a curriculum-style transfer learning pipeline: general chemical pretraining (Virtual Analogs), followed by metabolite-specific pretraining (MetaTrans), and fine-tuning on a rigorously curated dataset (LAGOM dataset) composed of DrugBank and MetXBioDB entries.
4.A key innovation is LAGOM’s single-model approach, which outperforms both the rule-based GLORYx and SyGMa tools and the previous Transformer-based MetaTrans model on the standard GLORYx benchmark dataset.
5.LAGOM incorporates SMILES randomisation during fine-tuning, which significantly boosts performance. Other augmentation strategies (e.g., parent-grandchild reactions, property annotations) had limited or even negative effects.
6.The curated LAGOM dataset includes over 4000 parent-metabolite pairs with strict filtering based on atom types, molecular weight, and Tanimoto similarity to ensure quality and eliminate overlaps with test sets.
7.LAGOM achieves superior precision (0.18 vs 0.11) and F1 score (0.25 vs 0.17) over MetaTrans, while maintaining comparable recall. This reflects better balance between identifying correct metabolites and avoiding false positives.
8.An ensemble approach using multiple LAGOM models trained on different data splits further improves recall, though precision tends to drop due to increased prediction diversity.
9.Evaluation metrics go beyond simple accuracy, focusing on recall, precision, and F1 score using top-k predictions per drug. All models maintain over 95% validity of generated SMILES strings.
10.This work also contributes a reproducible data curation and training pipeline, enabling future research in metabolite prediction using chemical language models.
11.Despite advances, the authors acknowledge that low-data regimes, high chemical diversity, and the one-to-many nature of metabolism remain challenges for model generalisation and evaluation.
12.Future directions include expanding the dataset with richer metabolic transformations and exploring better model selection strategies that reflect external benchmark performance.
💻Code: github.com/tsofiac/LAGOM
📜Paper: doi.org/10.26434/chemrxiv-20…
#DrugDiscovery #Chemoinformatics #AI4Science #MetabolitePrediction #TransformerModel #ComputationalPharmacology #SMILES #DeepLearning
2
18
1,202