
19 Nov 2025
Rationalized All-Atom Protein Design with Unified Multi-Modal Bayesian Flow
1. This groundbreaking study introduces ProBayes, a novel approach to all-atom protein design using Bayesian flows. It addresses a critical challenge in protein engineering by unifying the design of backbone structures, sequences, and side-chains in a single framework, significantly improving the accuracy and efficiency of protein generation.
2. The core innovation lies in the rationalized information flow strategy, which eliminates the “information shortcut” problem. This issue has plagued previous models, where sequence information could be inferred from side-chain data, compromising the model’s ability to learn true sequence distributions. ProBayes avoids this by carefully designing the information flow between different protein components.
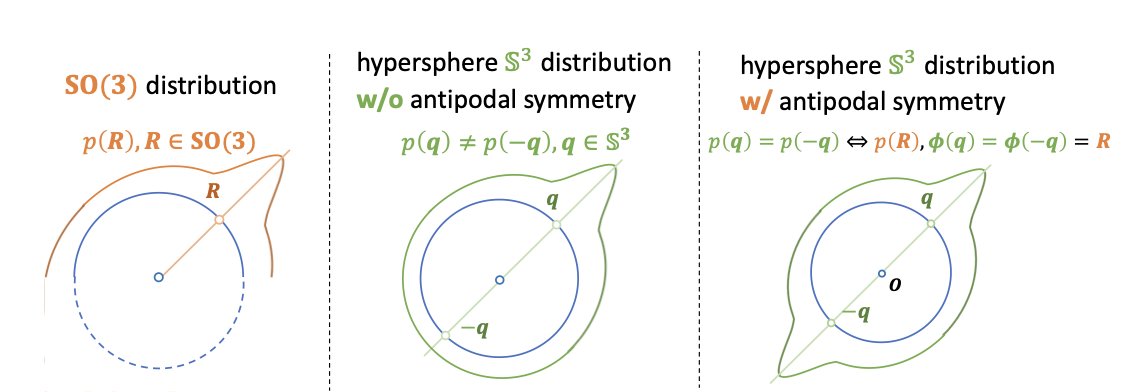
3. Another major contribution is the development of the first Bayesian flow formulation for protein backbone orientations. By transforming the complex SO(3) orientation modeling into an equivalent hyperspherical generation problem with antipodal symmetry, the authors bypass the difficulties of directly applying Bayesian inference on SO(3) matrices, enabling more accurate and efficient protein backbone generation.
4. ProBayes demonstrates exceptional performance in both peptide and antibody design tasks. For instance, it achieves a DockQ score of 0.74 on PepBench, significantly outperforming existing methods. The model also shows superior results in antibody design, achieving state-of-the-art metrics such as lower energy scores and higher sequence recovery rates.
5. The study provides a comprehensive validation through extensive experiments and ablation studies. Results show that ProBayes not only improves the quality of generated proteins but also maintains high computational efficiency. The rationalized information flow and Bayesian flow framework are proven to be effective in capturing the intricate relationships between different protein components.
6. The work has significant implications for therapeutic discovery, enzyme engineering, and synthetic biology. By enabling more accurate and efficient protein design, ProBayes could pave the way for the development of novel functional proteins with potential applications in medicine and biotechnology.
💻Code: github.com/GenSI-THUAIR/ProB…
📜Paper: openreview.net/pdf/5427b0c44…
#ProteinDesign #BayesianFlow #ComputationalBiology #AIforBiology
6
38
2,566
13 May 2025
Piloting Structure-Based Drug Design via Modality-Specific Optimal Schedule
1.Structure-based drug design (SBDD) critically depends on accurately generating molecular geometries conditioned on protein pockets. Yet, deep generative models often fail to model the interdependence between 3D positions and 2D molecular topology.
2.This work introduces VLB-Optimal Scheduling (VOS), a principled strategy to design optimal noise schedules in multi-modality generation, maximizing the Variational Lower Bound (VLB) and improving molecular geometry and binding accuracy.
3.Key insight: In multi-modality generative models, VLB becomes a path integral, meaning it is dependent on the full trajectory of noise schedules—not just the endpoints. This breaks the common assumption from single-modality diffusion models.
4.The authors formalize the search for an optimal probability path in the joint noise schedule space and reduce the search complexity to a 2D rescaled time grid, allowing for efficient optimization via dynamic programming.
5.By training a single model with a generalized loss objective that spans the entire schedule space, they enable inference-time interpolation and extrapolation of noise schedules—eliminating the need for retraining.
6.The resulting model, MolPilot, achieves a state-of-the-art PoseBusters passing rate of 95.9% on CrossDock and maintains 79.1% on the challenging PoseBusters OOD benchmark, outperforming all tested baselines.
7.MolPilot also delivers superior binding pose quality, with 56.1% of generated molecules matching redocking poses within 2Å RMSD, close to the ground-truth redocking upper bound of 59.4%.
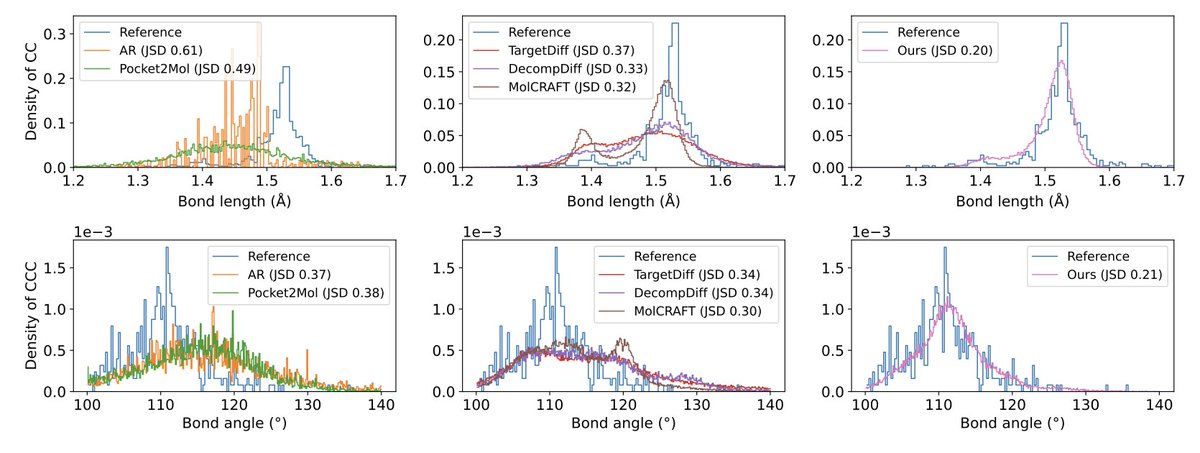
8.The method improves geometric accuracy, yielding better bond length and angle distributions (JSD scores), and higher structural validity in both in-distribution and out-of-distribution scenarios.
9.Ablation studies confirm both the generalized loss and optimal schedule independently and jointly contribute to improved VLB, better molecular conformations, and stronger protein-ligand interactions.
10.VOS is shown to be broadly applicable: integrating it into the diffusion-based TargetDiff framework yields significant improvements in pose quality and energy scores, demonstrating its flexibility beyond MolPilot.
11.This work not only sets a new benchmark for SBDD performance, but also proposes a theoretical and computationally efficient framework for future multi-modal generative modeling in chemistry and biology.
📜Paper: arxiv.org/abs/2505.07286
#MolecularGeneration #SBDD #MachineLearning #DrugDiscovery #GenerativeModels #DiffusionModels #BayesianFlow #GeometryLearning
2
3
1,744
13 May 2025
Piloting Structure-Based Drug Design via Modality-Specific Optimal Schedule
1.Structure-based drug design (SBDD) critically depends on accurately generating molecular geometries conditioned on protein pockets. Yet, deep generative models often fail to model the interdependence between 3D positions and 2D molecular topology.
2.This work introduces VLB-Optimal Scheduling (VOS), a principled strategy to design optimal noise schedules in multi-modality generation, maximizing the Variational Lower Bound (VLB) and improving molecular geometry and binding accuracy.
3.Key insight: In multi-modality generative models, VLB becomes a path integral, meaning it is dependent on the full trajectory of noise schedules—not just the endpoints. This breaks the common assumption from single-modality diffusion models.
4.The authors formalize the search for an optimal probability path in the joint noise schedule space and reduce the search complexity to a 2D rescaled time grid, allowing for efficient optimization via dynamic programming.
5.By training a single model with a generalized loss objective that spans the entire schedule space, they enable inference-time interpolation and extrapolation of noise schedules—eliminating the need for retraining.
6.The resulting model, MolPilot, achieves a state-of-the-art PoseBusters passing rate of 95.9% on CrossDock and maintains 79.1% on the challenging PoseBusters OOD benchmark, outperforming all tested baselines.
7.MolPilot also delivers superior binding pose quality, with 56.1% of generated molecules matching redocking poses within 2Å RMSD, close to the ground-truth redocking upper bound of 59.4%.
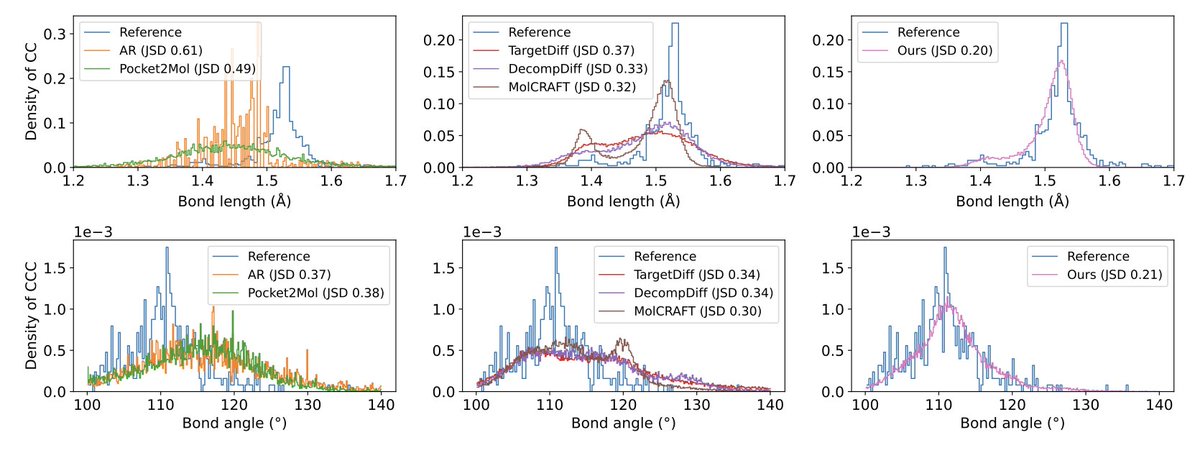
8.The method improves geometric accuracy, yielding better bond length and angle distributions (JSD scores), and higher structural validity in both in-distribution and out-of-distribution scenarios.
9.Ablation studies confirm both the generalized loss and optimal schedule independently and jointly contribute to improved VLB, better molecular conformations, and stronger protein-ligand interactions.
10.VOS is shown to be broadly applicable: integrating it into the diffusion-based TargetDiff framework yields significant improvements in pose quality and energy scores, demonstrating its flexibility beyond MolPilot.
11.This work not only sets a new benchmark for SBDD performance, but also proposes a theoretical and computationally efficient framework for future multi-modal generative modeling in chemistry and biology.
📜Paper: arxiv.org/abs/2505.07286
#MolecularGeneration #SBDD #MachineLearning #DrugDiscovery #GenerativeModels #DiffusionModels #BayesianFlow #GeometryLearning
2
2
8
2,336

