
The antibiotic pipeline has been stalled for decades. Discovering a new antibiotic takes 10 to 15 years and costs billions, and bacteria evolve resistance faster than that timeline allows. In June 2026, Nature mapped out what AI is doing to that problem, and one of the tools is mining the genomes of extinct animals for molecules that bacteria have never seen before.
The pipeline problem is structural. After decades of easy wins with natural compounds and simple chemistry, the low hanging fruit was gone by the 1980s. Drug-resistant infections directly killed more than 1.1 million people in 2021 according to The Lancet, a figure that is projected to rise steeply , and the WHO lists dozens of bacterial strains with no reliable treatment. The pharmaceutical industry largely walked away from antibiotic development because the economics don't work: a drug you take for ten days generates less revenue than a drug you take for life. Discovery fell behind resistance.
A Nature technology feature published June 8, 2026 mapped three distinct ways AI is beginning to compress that gap.
The first is molecular docking. A tool called DiffDock, developed in Regina Barzilay's lab at MIT, predicts with high accuracy how a drug candidate binds to its target protein. This matters because one of the most expensive steps in antibiotic discovery is figuring out exactly what a candidate molecule is doing and why it works. DiffDock can narrow that down computationally in hours rather than months, helping researchers quickly identify which candidates are worth pursuing and why.
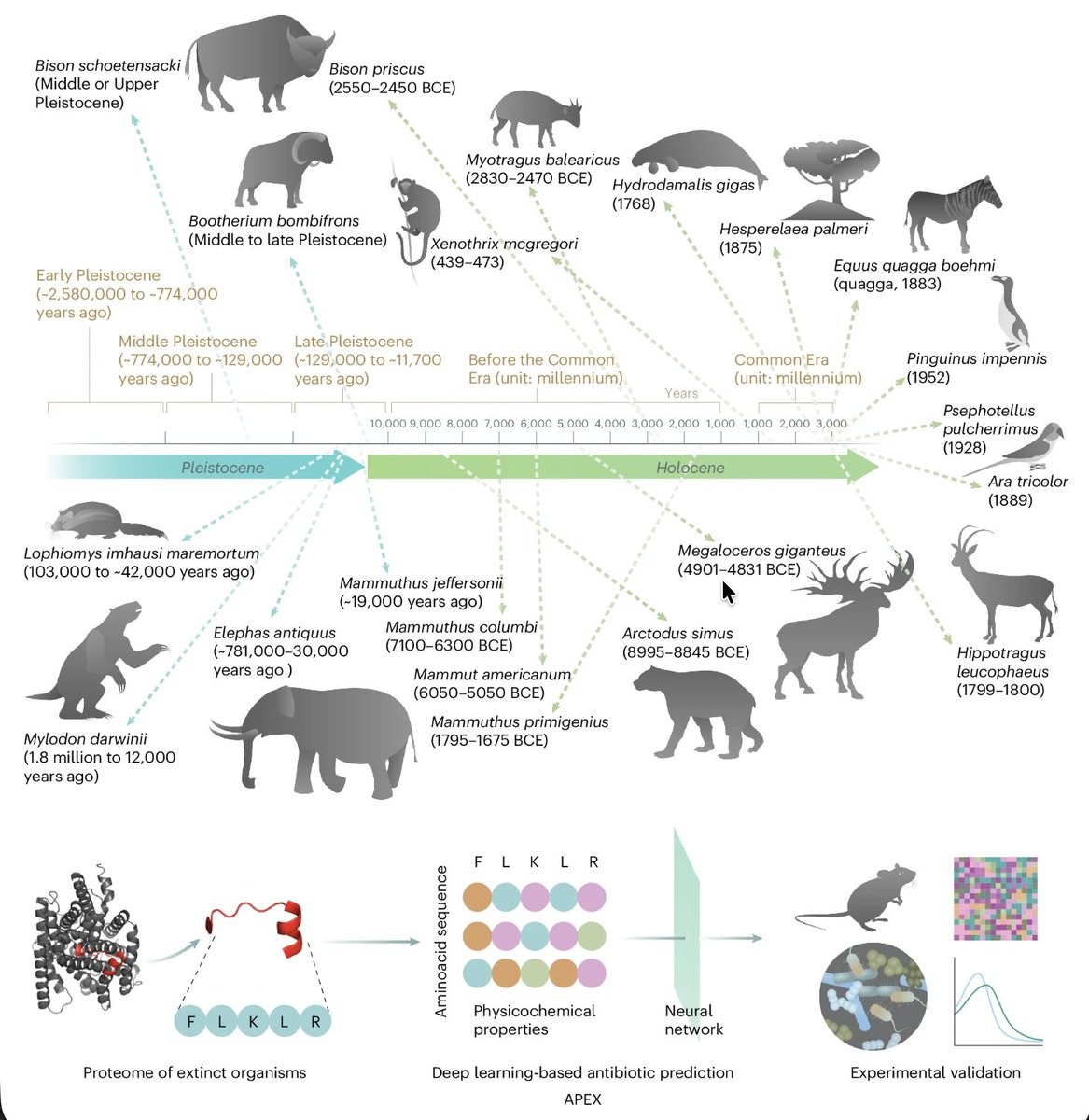
The second is molecular de-extinction. Cesar de la Fuente's lab at the University of Pennsylvania built a neural network system called APEX (Antibiotic Peptide de-EXtinction) that mines the protein sequences of extinct organisms. The logic is precise: bacteria evolve resistance to molecules they have encountered before. Molecules from organisms that have been gone for tens of thousands of years, like giant ground sloths, ancient magnolias, and Grant's zebras, are molecules living bacteria have never been exposed to. APEX screened more than ten million peptide sequences and identified over 37,000 candidates with predicted broad-spectrum antimicrobial activity, around 11,000 of them derived from extinct species. When 69 of these candidates were synthesized and tested in the lab, many showed an unusual mechanism of action, targeting the inner cytoplasmic membrane of bacteria rather than the outer cell wall, a strategy that may be harder for bacteria to evolve resistance against.
The third is generative design: using AI not just to find existing molecules but to design new ones that don't exist in nature, optimizing for activity, safety, and resistance evasion simultaneously.
The honest caveat is the same one that applies to all early stage drug discovery. Finding a promising candidate is the beginning, not the end. Clinical translation, regulatory approval, manufacturing, and funding remain long and expensive. The Nature feature notes that public and philanthropic funding are likely necessary because market economics still don't favor antibiotic development at the pace needed.
But the shift in the starting point is real. Where antibiotic hunting once meant grinding through soil samples and chemical libraries, it can now begin with a computational sweep across the genomes of everything that has ever lived.
Source: Nature, June 8, 2026. Technology feature "AI is taking on antibiotic resistance: here's how." nature.com/articles/d41586-0…. Reporter: Jyoti Madhusoodanan. Key tools: DiffDock (Barzilay lab, MIT); APEX and APEX-GO (de la Fuente lab, University of Pennsylvania). APEX paper: Torres et al., Nature Machine Intelligence, 2026.
11
15
1,458

AI is taking on antibiotic resistance — here’s how
AIが抗菌薬耐性に挑む――その方法
抗菌薬耐性により、かつて有効だった治療の信頼性が下がる一方で、新しい抗菌薬の発見は時間も費用もかかり、資金を集めにくい。
研究者はAIを使い、膨大な化合物の選別、有望な分子が細菌を攻撃する仕組みの予測、新しい抗菌ペプチドの設計を速めている。
例として、標的を絞った抗菌薬候補enterololinではDiffDockが作用機序の解明を助け、halicinは細菌増殖データで訓練したニューラルネットワークにより見いだされた。
別のチームは古代・絶滅生物由来のタンパク質を探り、生成AIで新分子を設計している。
課題は、質が高く多様なデータを整え、AIが設計した化合物を実際に作れるようにすることだ。
AI is helping researchers fight antibiotic resistance by screening molecules faster, predicting how drug candidates work and designing new antimicrobial peptides. Tools such as DiffDock, Chemprop and generative models are expanding discovery, but success depends on diverse, well-labelled data and on whether AI-designed compounds can be synthesized and tested.
#抗菌薬耐性
#AI創薬
#抗菌ペプチド
#AntibioticResistance
#AIDrugDiscovery
#AntimicrobialPeptides
nature.com/articles/d41586-0…
14 Oct 2025

WHO、世界で一般的な抗菌薬への広範な耐性を警告—2025年グローバル耐性監視報告
WHOの新たな監視報告は、抗菌薬耐性が世界的に広がり増加していると警告する。
2023年には検査で確認された感染の6件に1件が治療薬に耐性を示した。
2018〜2023年に監視対象の病原体と抗菌薬の組合せの4割超で耐性が上昇し、年5〜15%増だった。
22種類の抗菌薬と8病原体の推定では、東南アジア地域と東地中海地域で約3件に1件、アフリカ地域で5件に1件が耐性だった。
特にグラム陰性菌(E. coli、K. pneumoniae)の血流感染で第三世代セファロスポリン耐性が高く、アフリカ地域では70%超。
WHOは2030年までのGLASS高品質報告とワンヘルス対策の強化を求める。
WHO reports rapid, global growth of antibiotic resistance: one in six infections was drug-resistant in 2023. Gram-negative pathogens, notably E. coli and K. pneumoniae, show high resistance to third-generation cephalosporins, limiting treatments. Surveillance has expanded but remains patchy. WHO calls for robust GLASS data and coordinated One Health measures by 2030.
who.int/news/item/13-10-2025…
1
3
407

DiffDock-Glide: A Hybrid Physics-Based and Data-Driven Approach to Molecular Docking #machinelearning #compchem pubs.acs.org/doi/abs/10.1021…
5
10
493

May 18
Because molecular generation is the trap.
The industry assumes we lack the right molecules. We don't. We lack the causal map of the molecules we already have.
There are thousands of clinically safe, FDA-approved, and GRAS compounds. But their downstream network effects and how they act in multi-compound matrices to modulate complex diseases are largely unmapped by human silos.
When you use AI to hallucinate a novel molecule, you buy a 10-year, $1B ticket through Phase 1 toxicity trials. When you use deterministic AI to mathematically map the topology of *existing* safe compounds against clinical anomalies, you bypass Phase 1 entirely.
I am not proposing a screening layer for de novo molecules. I am saying de novo generation is an ego-driven waste of capital.
To simplify without giving away the architecture: we are doing things the hard way because the industry is obsessed with complexifying knowledge extraction. The establishment thinks 'AI drug discovery' means training massive Graph Neural Networks (GNNs) or diffusion models (like DiffDock) to hallucinate geometry in a vacuum. You throw 100 million compounds into a latent space and pray the vector math spits out a cure. That is just hyper-expensive statistical guessing.
If I actually needed to generate a novel molecule, I wouldn't start by hallucinating geometry. I would take the existing dataset of approved molecules, extract the exact physical constraints that produce their clinical effects (the isomorphic mechanism), and use the AI purely to map a structure that satisfies those strict constraints.
My point is: the way we extract information is fundamentally flawed. You don't use LLMs to *generate*. You use them to *compute causality*.
Current methods treat AI like a hyper-advanced search engine. Sovereign architecture treats AI like a constraint-satisfaction solver. We have enough existing biological ground-truth to solve these diseases right now, if we stop overcomplicating how we query it.
1
3
42

May 15
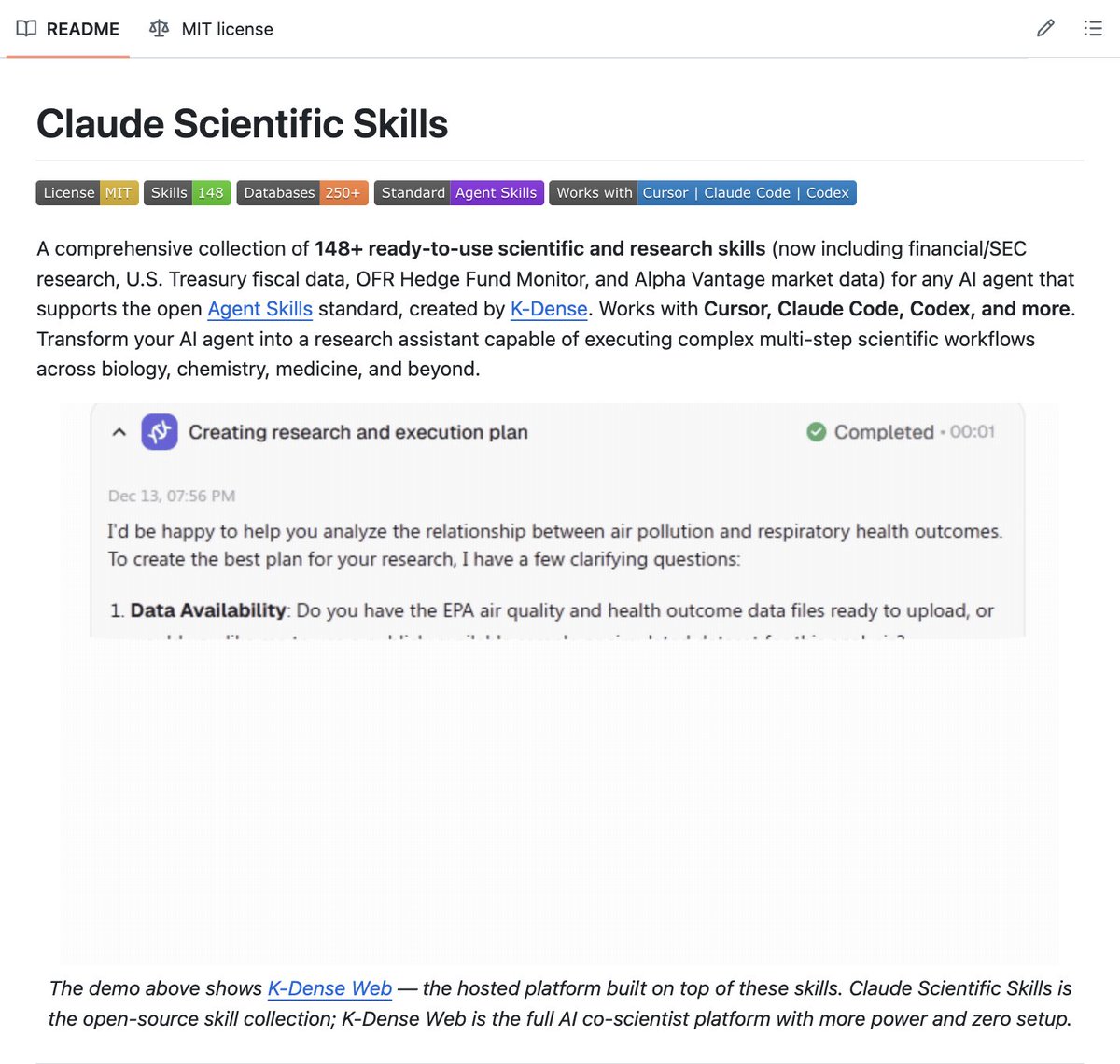
🚨 Someone just open sourced 135 ready-to-use skills that turn Claude Code into a full AI scientist.
Cancer genomics. Drug discovery. Molecular dynamics. Protein folding. RNA analysis. Geospatial science. Time series forecasting. 78 scientific databases. All accessible from a single command in your terminal.
It's called Scientific Agent Skills. Built by K-Dense. And it works with Claude Code, Cursor, Codex, and Gemini CLI out of the box.
Here's what was missing.
Claude Code is powerful. But when a researcher asks it to "find allosteric modulators for this protein-protein interaction," it doesn't know about AlphaFold DB, ZINC, DiffDock, DeepChem, or USPTO patents. It knows how to code. It doesn't know the scientific infrastructure that research actually runs on.
Scientific Agent Skills teaches it.
Each skill is a structured SKILL.md file that tells the agent exactly how to use a specific scientific tool, database, or package. The agent reads the skill automatically when it's relevant. No prompting required. No explaining what PubMed is every session.
Here's what's inside:
→ 30 scientific and financial databases — PubMed, bioRxiv, ChEMBL, UniProt, COSMIC, ClinicalTrials.gov, SEC EDGAR, Alpha Vantage, OpenAlex, and more
→ 55 Python packages — RDKit, Scanpy, BioPython, PyTorch Lightning, PennyLane, Qiskit, and others with full usage patterns
→ 15 scientific integrations — Benchling, DNAnexus, LatchBio, OMERO, Protocols.io
→ GPU acceleration frameworks — CuPy, Numba, cuML, cuGraph, cuDF, all with decision frameworks for when to use each
→ Lab automation — PyLabRobot for controlling liquid handling robots, plate readers, incubators
→ Cancer genomics, drug-target binding, molecular dynamics, RNA velocity, geospatial science, time series forecasting
Here's how it works in practice.
You ask your agent to discover allosteric modulators for a protein-protein interaction. It automatically retrieves AlphaFold structures, identifies interaction interfaces with BioPython, searches ZINC for candidates, filters with RDKit, docks with DiffDock, ranks with DeepChem, checks PubChem suppliers, and searches USPTO patents.
End to end. From one research question.
Here's the wildest part.
Your AI agent discovers the skills automatically and uses them when relevant. Just a code. No configuration. No explaining the tools. No teaching your agent what databases exist.
K-Dense also released BYOK — a free desktop AI co-scientist powered by these skills that works with 40 models, runs locally, and includes web search, file handling, and optional cloud compute via Modal.
2.4K forks. 135 skills. Actively maintained by K-Dense.
100% Open Source. MIT License.
GitHub link in the comments 👇
2
6
8
457

Fine-Tuning DiffDock-L for Allosteric Kinase Docking #Allostery #Docking
pubs.acs.org/doi/10.1021/acs…
#JCIM Vol66 Issue6 #MachineLearning #DeepLearning
5
26
1,405

Accelerating Drug Discovery with HyperLab: An Easy-to-Use AI-Driven Platform
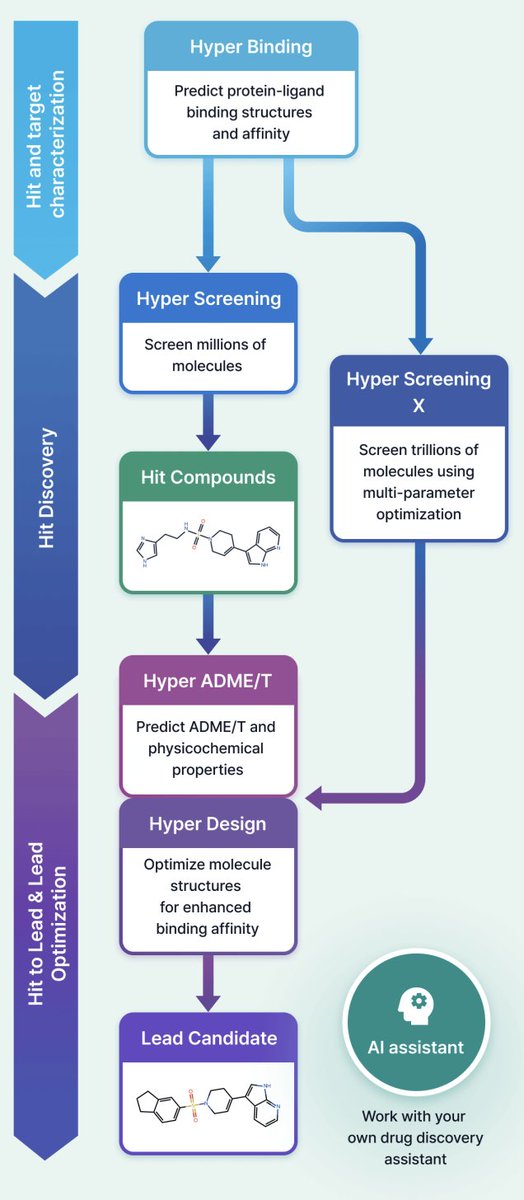
1 HyperLab (by HITS) is presented as a web-based, AI-driven SBDD platform aimed at making structure-based workflows usable by experimental drug discovery researchers without requiring AI/CADD expertise, emphasizing integrated UI/UX over fragmented toolchains.
2 The platform compresses early discovery into a single environment spanning: protein–ligand pose affinity prediction (Hyper Binding), covalent complex modeling (Covalent Hyper Binding), virtual screening from 1M to 11T compounds (Hyper Screening / Hyper Screening X), structure-based molecular optimization (Hyper Design), SAR analysis, and 19-endpoint ADME/T prediction (Hyper ADME/T), with an embedded AI assistant for workflow automation.
3 Hyper Binding’s key technical angle is physics-informed deep learning for protein–ligand interactions, supporting multiple protein inputs (PDB ID, uploaded PDB, AlphaFold structures via UniProt) and an end-to-end co-folding mode that predicts complex structures directly from protein sequence plus ligand, reducing dependence on curated receptor structures.
4 On PoseBuster v2 (PB-valid) pose prediction, Hyper Binding reports 77% accuracy when given binding-site information, compared with 58% for Vina and 13% for DiffDock; it approaches AlphaFold3 (84%) and is comparable to Boltz2 (78). The paper also highlights throughput: ~3 minutes per complex (via cloud) vs ~15 minutes for AlphaFold3 on an RTX 3060.
5 For binding affinity prediction on two FEP-style benchmarks (focused on subtle potency differences among close analogs), Hyper Binding reports Pearson r = 0.70 and 0.53, outperforming evaluated deep learning scorers (Luminet, GenScore) and physics-based docking (Glide SP, Vina) on both datasets.
6 Covalent drug discovery is treated as a first-class workflow: covalent pose prediction is benchmarked on a curated covalent set (from PDBBind/PDB). Covalent Hyper Binding (cofolding) reports 88.7% pose accuracy vs 48.4% (COV SMINA) and 46.8% (GNINA); the docking mode reports 61.3%. Screening enrichment (EF@10%) is reported as 6.56 (Mpro) and 9.97 (KRAS), exceeding baselines under the described setup.
7 Hyper Screening targets rapid hit finding by running Hyper Binding across curated libraries and returning top-ranked candidates (top 500). Built-in libraries include: Diverse (1,000,000), Fragment (500,000; rule-of-three-like), Kinase-focused (65,000), Natural product-like fragments (4,200), and FDA-approved (1,100), plus support for user-registered libraries.
8 Hyper Screening X expands to an 11-trillion-molecule virtual space using generative exploration with GFlowNet-based models, optimizing binding score plus properties (e.g., MW, TPSA, LogP). The workflow is described as: set target property constraints, train (~48h), then generate molecules (e.g., 100 molecules in ~30 min), with synthetic route output and optional synthesis request via a partner service.
9 Hyper Design provides structure-based optimization starting from a scaffold or an X-ray-bound ligand, enabling user-specified modification sites and fragment growth/replacement with synthesizability constraints; outputs include 3D structures and iterative “design trees.” The paper positions use cases as fragment-to-lead growth and generating patent-distinct analogs while preserving key interactions.
10 The internal validation study emphasizes “no post-analysis/visual inspection” selection: a 24-hour Hyper Screening run led to 52 compounds tested, yielding 5 hits with IC50 70–600 nM (~9% hit rate). Hyper Design then produced derivatives; 5 were synthesized and 3 showed >75% inhibition at 1 µM with IC50 200–400 nM, including one compound comparable or better than a reference and with supporting pathway assay readouts.
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #ComputationalBiology #Cheminformatics #StructureBasedDrugDesign #VirtualScreening #CovalentInhibitors #ADMET #GenerativeAI #ProteinLigandDocking #BioRxiv
3
16
1,463

Apr 1
Drug discovery is broken at the design stage!
Not because the biology is too hard, but because the tools are fragmented, the iteration cycles are too long, and too much is still done by instinct rather than computation.
At Boltzmann Labs, we've built an end-to-end small molecule discovery platform that changes this.
From a single target, our platform takes you through:
→ De novo molecule generation — structure-based, pharmacophore-based, and generative graph models
→ Intelligent screening — ADMET, toxicophore, novelty (vs. ChEMBL SureChemBL), similarity & substructure filters
→ Custom QSAR modelling — train predictive models on your own activity data using AutoML, DNN, Random Forest, and more
→ Binding site prediction & molecular docking — AutoDock Vina, DiffDock, EquiDock, flexible docking
→ MD simulation — OpenMM and GROMACS, fully configurable
→ Multi-objective lead optimization — Pareto front, Graph GA, MARS Markov sampling, fragment-based optimization
The result: fewer wasted synthesis cycles, earlier go/no-go decisions, and a dramatically compressed path from target to candidate.
We're actively looking to partner with pharma and biotech teams who want to accelerate their early-stage discovery programs, and investors who see the compounding value of AI-native drug discovery infrastructure.
If that's you, let's talk. Reach out directly or drop a comment below.
1
4
118

🚨 Funding alert for computational chemists & drug discovery researchers! Up to $5,000 to compare ML-based (Boltz-2, DiffDock…) vs. classical (AutoDock Vina, DOCK 6…) in silico screening methods on a target of your choice — open to applicants worldwide, any career stage.
4
13
81
9,419
Teaching Diffusion Models Physics: Reinforcement Learning for Physically Valid Diffusion-Based Docking
1. The paper frames a key failure mode of diffusion docking: models can hit “near-native” RMSD thresholds while still producing steric clashes and missing critical protein–ligand interactions, showing that RMSD alone is misaligned with practical pose quality.
2. They introduce an RL fine-tuning framework for diffusion-based docking that directly optimizes non-differentiable downstream objectives, using PoseBusters physical validity checks (and proximity to the crystal pose) as the terminal reward—without adding any inference-time compute.
3. Methodologically, they cast reverse diffusion as an MDP (each denoising step is an action; stochasticity comes from the policy), and optimize the diffusion policy with a DDPO-style clipped policy-gradient objective with per-complex reward normalization.
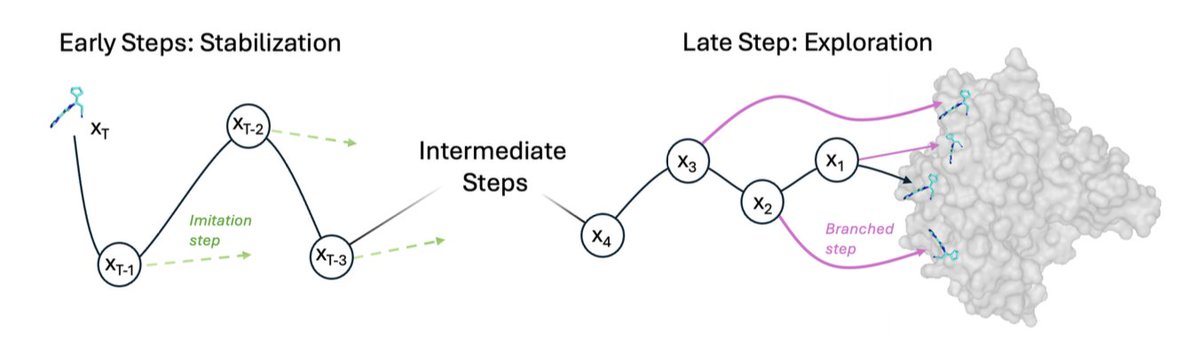
4. Innovation 1: early-step imitation regularization. Because terminal rewards make credit assignment noisy, early high-noise denoising steps are regularized toward “expert” actions that move translation/rotation/torsions toward the ground-truth pose, stabilizing training while leaving later steps to RL.
5. Innovation 2: late-step trajectory branching. Near the end of denoising (t in {8,…,5}), they branch trajectories by resampling noise to produce 16 leaf poses per rollout, then assign rewards through the tree (shared prefixes get averaged descendant rewards). This increases sample efficiency and sharpens learning around validity boundaries (e.g., clash vs no-clash).
6. Results on PoseBusters (308 complexes) show large gains in physical plausibility: PB-validity for the Top-1 pose rises from 58.8% to 78.1%, and across all sampled poses from 38.2% to 58.9%. The biggest jump is on out-of-distribution targets (0–30% sequence identity): PB-valid samples increase from 24.3% to 46.4%.
7. Importantly, physical validity improves without sacrificing accuracy: Top-1 success for the joint criterion RMSD ≤2Å & PB-valid improves from 46.2% to 58.8%; Oracle success from 66.1% to 79.9%. Interaction recovery also improves despite not being explicitly optimized (e.g., Top-1 with ≥50% IR rises 38.9% to 49.7%).
8. RL shifts the generated pose distribution toward more favorable physics-based energies: mean Vina energy across generated poses improves from 2.24 kcal/mol to −2.10 kcal/mol, consistent with fewer clashes and more energetically plausible conformations.
9. Compared with docking baselines, DiffDock-Pocket RL outperforms Vina and GOLD under stricter “useful pose” criteria that require validity and interaction recovery. With simple post-hoc minimization (smina/Vina) and GNINA re-ranking (“RL ”), Top-1 reaches 80.2% for RMSD ≤2Å and 78.2% for RMSD ≤2Å & PB-valid, exceeding all tested methods on PoseBusters.
10. Analysis suggests RL fixes two common baseline failures: (a) near-native poses that are physically invalid (clashes), and (b) failing to reach the native binding mode at all. RL also increases the diversity of PB-valid poses (higher mean pairwise RMSD among valid samples), supporting improved exploration rather than memorization.
💻Code: github.com/oxpig/RLDiff
📜Paper: biorxiv.org/content/10.64898…
#DiffusionModels #ReinforcementLearning #MolecularDocking #ComputationalChemistry #DrugDiscovery #StructuralBiology #GenerativeAI #MachineLearning
9
97
5,508
Sigmadock: Untwisting Molecular Docking with Fragment‑Based SE(3) Diffusion
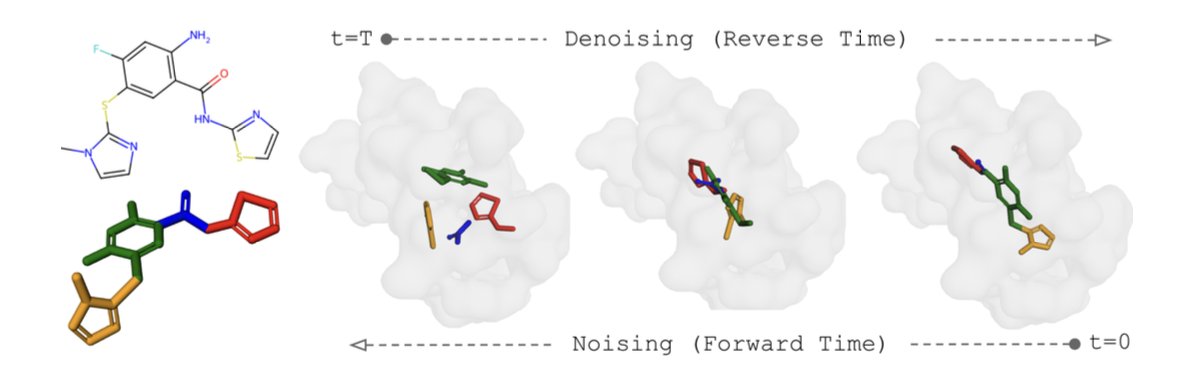
1 This paper presents Sigmadock, the first diffusion‑based docking model that surpasses classical physics‑based tools on the PoseBusters re‑docking benchmark, achieving a Top‑1 success rate over 79 % under the strict PB‑validity metric.
2 The core innovation is a novel fragmentation scheme that breaks a ligand into a small set of rigid‑body fragments by cutting rotatable bonds, allowing the model to learn only SE(3) transformations for each fragment rather than high‑dimensional torsional angles.
3 By operating in the product space SE(3)^m, Sigmadock sidesteps the geometric entanglement that plagues torsional‑space diffusion—where a single dihedral change propagates non‑locally and induces a non‑product measure—leading to more stable training and faster inference.
4 The authors further introduce soft triangulation constraints that enforce bond‑length and angle consistency across fragments, and a SO(3)‑equivariant EquiformerV2 backbone that respects the rotational symmetry of the 3‑D space.
5 Extensive ablation studies show that each component—fragment merging, triangulation, and protein‑ligand interaction encoding—contributes 4–12 % to overall accuracy, and the method generalises to unseen proteins with little data leakage.
6 On the PoseBusters and Astex test sets, Sigmadock reaches near‑perfect Top‑1 accuracy (>90 %) and outperforms both DiffDock and traditional docking programs by large margins, all while using only ~19 k training molecules and 50× faster sampling.
7 The work demonstrates that principled inductive biases and careful geometric modeling can enable deep learning to reliably predict binding poses, opening the door to flexible‑receptor docking and co‑folding extensions.
💻Code: github.com/alvaroprat97/sigm…
📜Paper: arxiv.org/abs/2511.04854
#DeepLearning #MolecularDocking #DiffusionModels #ComputationalChemistry #SE3 #FragmentBasedApproach
3
21
88
12,307

Mar 20
SandboxAQ is thrilled to celebrate the incredible innovations that were showcased at NVIDIA GTC!
At SandboxAQ, we are proud to highlight our partnership with NVIDIA, which is accelerating the future of AI in life sciences, drug discovery, and beyond. By combining SandboxAQ's Large Quantitative Models (LQMs) with NVIDIA's DGX Cloud, we are tackling some of the world's most computationally intensive scientific challenges.
Here are a few major milestones from our collaboration:
🧬 The SAIR Dataset: We jointly released the Structurally-Augmented IC50 Repository (SAIR), the world's largest open dataset of co-folded 3D protein-ligand structures paired with experimental binding affinity labels. Featuring over 5.2 million structures, this massive dataset was generated using a managed Kubernetes environment powered by 768 NVIDIA H100 GPUs. Thanks to this computing power, we compressed months of compute time into just three weeks—a 4x speedup!
🔬 OpenFold3: As a key contributor to the OpenFold Consortium, we’ve already built on earlier OpenFold advances in our AQAffinity model for binding-affinity prediction, and we’re now adopting OpenFold3 to surcharge those capabilities even further.
🧪 AQAffinity: Our sequence-to-affinity model, joining the OpenFold Consortium to enable key drug discovery workflows.
💊 DiffDock NIM: NVIDIA has leveraged our SAIR dataset to significantly increase the accuracy of their DiffDock NIM, a state-of-the-art generative diffusion model for molecular blind docking, which is now available on the NVIDIA build portal.
⚡ AQCat25 Dataset: To tackle complex challenges in heterogeneous catalysis, we generated an unprecedented spin-aware dataset of 13.5 million high-fidelity DFT calculations, powered entirely by an NVIDIA DGX cluster.
With four co-authored scientific papers, several thought leadership webinars, and a newly formed NVIDIA-SandboxAQ joint steering committee, our collaboration is stronger than ever.

4
8
398

The most important Claude plugin in existence just dropped on GitHub and nobody is talking about it.
Follow for more AI content. @USHirshwar
Install it once. Claude becomes a full AI research scientist permanently.
Here's what it can run from a single prompt:
→ Full drug discovery pipelines with real bioactive compound queries
→ Single-cell RNA sequencing analysis with Scanpy
→ Clinical variant annotation with ClinVar and Ensembl
→ Molecular docking against AlphaFold structures via DiffDock
→ Patient-to-trial matching via live ClinicalTrials.gov data
→ Publication-ready PDF clinical reports generated automatically
Bioinformatics. Cheminformatics. Proteomics. Quantum computing. Medical imaging. Laboratory automation.
All connected to the databases and tools scientists actually use.
One prompt. Real science. Actual results.
This is not a chatbot anymore.
/plugin install scientific-skills@claude-scientific-skills
100% open source. MIT License.

1
1
2
1,295

Mar 5
🚨 BREAKING: The most important Claude plugin in existence just dropped on GitHub and nobody is talking about it.
It's called claude-scientific-skills.
140 scientific skills across every major research domain baked into one plugin.
Install it once. Claude becomes a full AI research scientist permanently.
Here's what it can run from a single prompt:
→ Full drug discovery pipelines with real bioactive compound queries
→ Single-cell RNA sequencing analysis with Scanpy
→ Clinical variant annotation with ClinVar and Ensembl
→ Molecular docking against AlphaFold structures via DiffDock
→ Patient to trial matching via live ClinicalTrials. gov data
→ Publication-ready PDF clinical reports generated automatically
Bioinformatics. Cheminformatics. Proteomics. Quantum computing. Medical imaging. Laboratory automation.
All connected to the databases and tools scientists actually use.
One prompt. Real science. Actual results.
This is not a chatbot anymore.
/plugin install scientific-skills@claude-scientific-skills
100% Open Source. MIT License.
Link in the comments.
43
279
1,556
122,217

Mar 3
🚨BREAKING: The "Cursor for scientific research" just dropped and it runs entirely inside Claude Code.
It's called claude-scientific-skills and it gives Claude 140 ready-to-use scientific skills from a single plugin install.
No API doc hunting. No library configuration. No duct-taped research pipelines.
It's powered by 28 live scientific databases wired directly into Claude.
→ Describe your research goal in plain English
→ Claude finds the right skill automatically
→ Full pipeline runs: data retrieval → analysis → publication-ready output
→ Works across biology, chemistry, medicine, ML, and clinical research
All running inside Claude Code. Zero manual setup.
But it's not just a prompt library.
It's a full AI research lab:
→ Drug discovery: ChEMBL → RDKit → DiffDock → lead optimization in one prompt
→ Genomics: 10X data → Scanpy → GRN inference → pathway enrichment
→ Clinical: VCF → ClinVar → pharmacogenomics → patient report
→ Multi-omics: RNA-seq proteomics metabolomics integrated automatically
7.8k stars. 924 forks. MIT Licensed.
MacOS, Windows, Linux works everywhere Claude Code runs.
This is the moment AI stops being a chat tool and becomes an actual research partner.
Link in the first comment 👇
25
137
872
79,077

Mar 1
🚨 Someone just turned Claude into a full AI research scientist.
This GitHub repo called claude-scientific-skills just quietly changed what's possible with AI in science.
Most people are using Claude to write emails and summarize docs.
These researchers are using it to run actual drug discovery pipelines, analyze single-cell RNA sequencing data, interpret clinical variants, and generate publication-ready reports all from a single prompt.
Here's how it works:
You install one plugin in Claude Code. Claude automatically discovers and uses 140 scientific skills across every major research domain bioinformatics, cheminformatics, proteomics, clinical research, medical imaging, materials science, quantum computing, laboratory automation.
The skills connect Claude directly to the databases and tools scientists actually use:
→ Query ChEMBL for bioactive compounds
→ Annotate variants with ClinVar and Ensembl
→ Dock molecules with DiffDock against AlphaFold structures
→ Analyze 10X genomics data with Scanpy
→ Search ClinicalTrials. gov and match patients to trials
→ Generate PDF clinical reports with ReportLab
One prompt. Real scientific libraries. Live database APIs. Actual results.
This is what happens when someone stops treating Claude like a chatbot and starts treating it like a research platform.
/plugin install scientific-skills@claude-scientific-skills
100% Opensource. MIT License.
(Link in the comments)
21
216
1,228
96,042
Feb 5
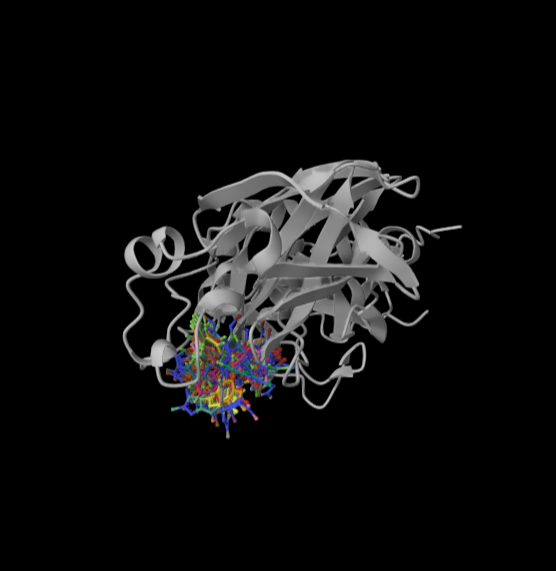
NVIDIA's DiffDock NIM now integrates SandboxAQ’s SAIR dataset, delivering consistent accuracy gains across every major docking metric on BioNeMo.
This is what happens when physics-based synthetic data meets high-quality experimental data. SAIR uniquely integrates physics-based modeling with key LQM capabilities, enabling improved generalization, enhanced reliability, and greater applicability for diverse drug discovery tasks. And when those models run on NVIDIA’s BioNeMo stack, the impact compounds.
If SAIR can materially improve DiffDock, imagine what SandboxAQ–hardened data and AI simulation tools could do for your models: sandboxaq.com/sair
1
3
350

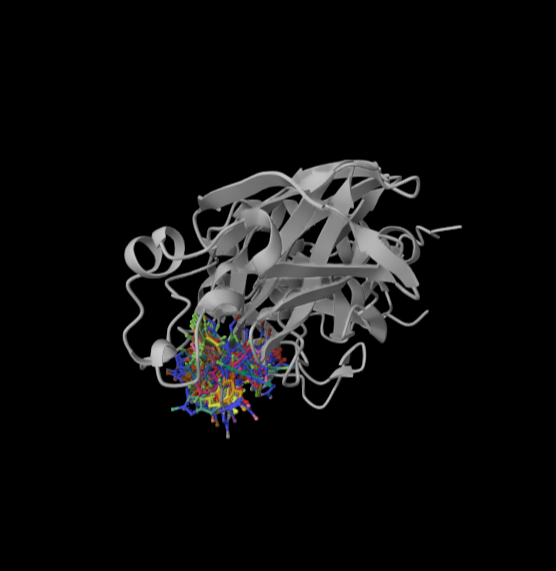
🚀 Smarter, faster docking on BioNeMo
The DiffDock NIM now integrates @SandboxAQ’s SAIR dataset, delivering consistent accuracy gains across all docking metrics.
A powerful proof that physics-based synthetic data experimental data = better AI for drug discovery 🧬
Try it 👇
4
16
113
5,743
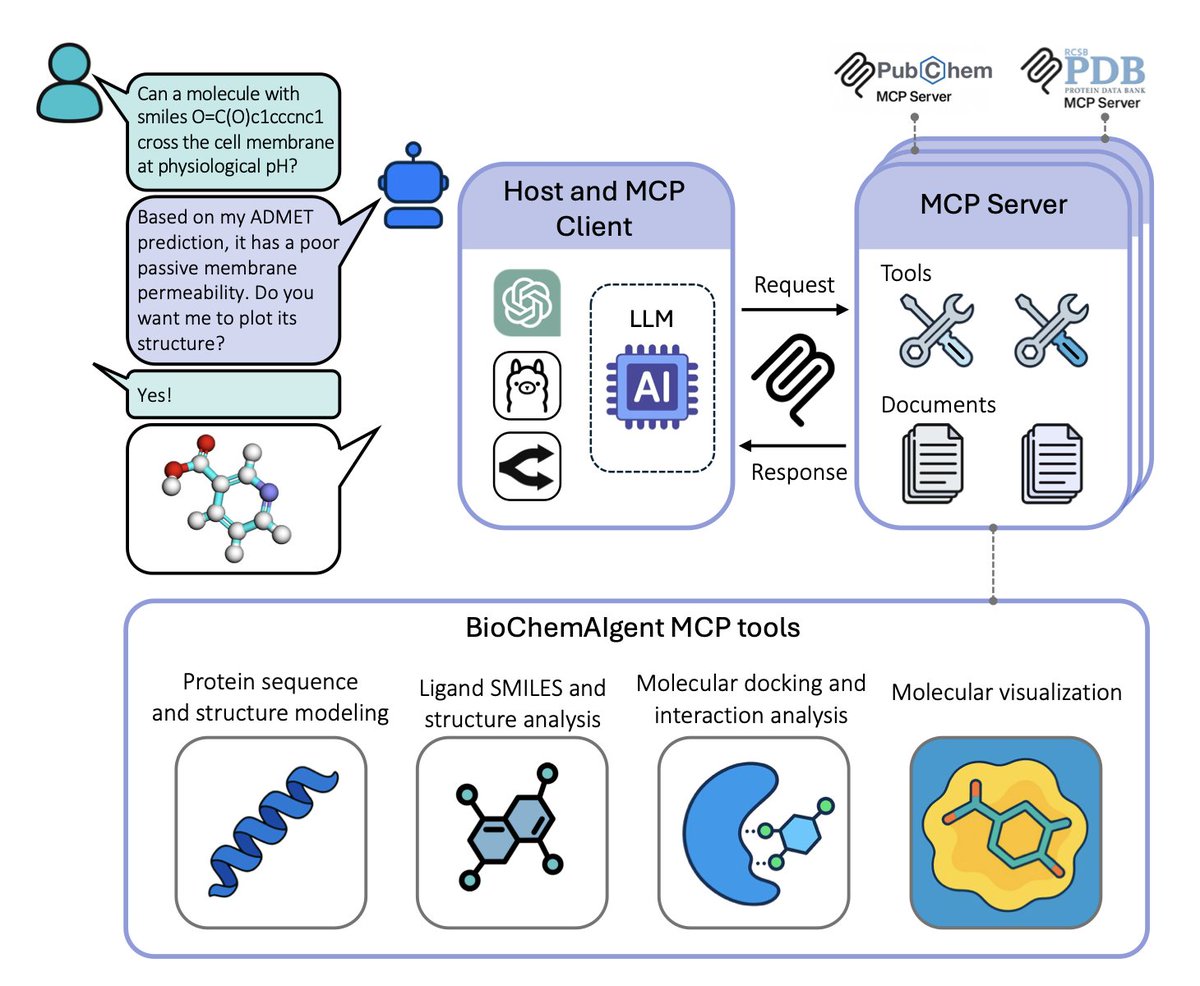
BioChemAIgent: An AI-driven Protein Modeling and Docking Framework for Structure-Based Drug Discovery
1. First agentic framework that unifies 19 tools—ESM3, AlphaFold3, AutoDock Vina, DiffDock, etc.—into one chat-style interface for end-to-end small-molecule discovery, slashing the usual multi-software integration burden.
2. Wraps complex pipelines (protein prep, protonation, grid setup, docking, interaction fingerprinting, 3-D visualization) into reproducible, transparent workflows that non-coders can launch with plain English.
3. Built-in “render_structures” and “interaction_plot” modules translate natural-language style requests into publication-grade 3-D images, eliminating the need for PyMOL scripting expertise.
4. Dual evaluation strategy: 98.5 % automatic accuracy on 65 corrupted queries plus perfect expert scores on four real-world case studies, showing GPT-5-powered agent rivals trained chemists in task planning and result interpretation.
5. Community-oriented registry on GitHub invites developers to plug in new MCP servers, turning the system into a living, expandable ecosystem rather than a frozen black box.
6. Open web interface already live—users can dock ibuprofen to COX-1, visualize the Arg120 salt bridge, and download inputs/outputs without installing anything.
💻Code: github.com/imsb-uke/bcai
📜Paper: biorxiv.org/content/10.64898…
#DrugDiscovery #AI #StructuralBiology #MolecularDocking #AlphaFold3 #ESM3 #ChemInformatics #OpenScience
12
50
2,773