
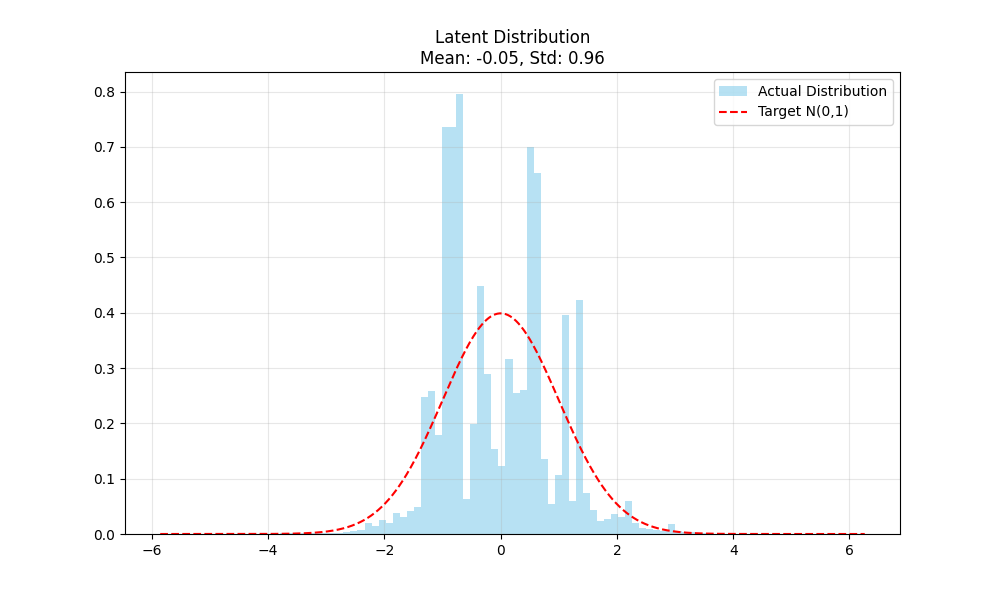
Jun 13
Open-source:
📄 arxiv.org/abs/2606.08670
💻 github.com/sisinflab/WaveDiT
Thanks to my coauthors Angela Lombardi, Giuseppe Fasano, @MattAttimonelli , @TommasoDiNoia at SisInfLab, and to @MICCAI_Society.
#MICCAI #MedicalImaging #GenerativeAI #FlowMatching #Wavelets #Neuroimaging #MRI
1
114

Jun 12
【物理現象の予測、生成AIが新しい解を導きます。】
👇 記事全文はこちら ecomottblog.com/?p=24195
#FlowMatching #生成AI #流体解析 #シミュレーション #ディープラーニング #デジタルツイン #エコモット
2
130

Flexible Flows for Biological Sequence Design
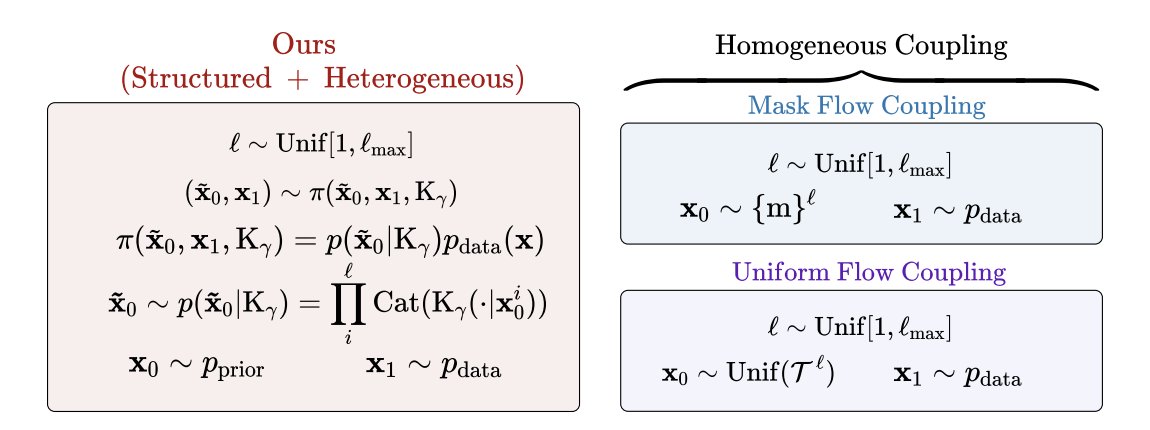
1. FlexFlow reframes discrete flow matching for biological sequences by changing the coupling (forward endpoint pairing) rather than the training objective: a structured, biology-informed coupling uses substitution matrices (e.g., BLOSUM for proteins; JC69/HKY85-style biases for nucleotides) to tilt the source distribution toward evolutionarily plausible neighborhoods.
2. The key idea is to keep the standard token-wise mixture path and CTMC machinery intact, but swap the usual “uninformative” couplings (uniform/masked) with a transition kernel Kγ that encodes preferred substitutions; when Kγ is uniform, the method reduces to the standard uniform coupling.
3. For variable-length generation, FlexFlow builds on Edit Flows by parameterizing reverse-time CTMC rates via edit operations (insertion, deletion, substitution). Instead of treating positions independently, it introduces a shared global latent r that conditions per-position edit decisions, coupling token-level operations through sequence-level context.
4. FlexFlow adds test-time control over edit behavior: operation probabilities are temperature-scaled and modeled with a Dirichlet prior over {ins, sub, del}. By changing Dirichlet concentrations α post-hoc, users can bias generation toward more insertions vs substitutions vs deletions without retraining, effectively acting as an “operation budget controller.”
5. The paper proposes latent classifier-free guidance (CFG) as an alternative to rate-space guidance: it performs CFG by interpolating conditional/unconditional latents (rc and r∅) in continuous space, then uses the guided latent to drive all edit operations jointly—aiming for more globally coherent conditioning than token-wise rate guidance.
6. The latent guidance has a probabilistic interpretation: under Gaussian conditional/unconditional latent encodings and sufficiency assumptions, the guidance direction corresponds to the score of an implicit classifier p(c|r), making the latent interpolation analogous to a gradient ascent step on log p(c|r).
7. Training uses an augmented alignment space with a blank token ε to make edit-based objectives tractable: alignments define edit sequences between endpoints, and a Bregman-divergence-style loss penalizes extraneous rates while rewarding edits that move xt toward x1.
8. DNA enhancer generation (unconditional, length 500) on fly brain and melanoma ATAC-seq datasets: FlexFlow achieves the best Fréchet Biological Distance among compared diffusion/flow baselines at the same sampling budget (100 reverse steps), and ablations indicate combining a frequency-informed prior with structured coupling performs best.
9. Conditional promoter design (human promoters, length 1024) conditioned on transcription initiation profiles: FlexFlow improves MSE of predicted regulatory activity versus prior baselines, with latent guidance outperforming rate guidance (reported 0.022 vs 0.024 MSE at 100 steps), suggesting benefits from global latent steering.
10. A new peptide–MHC II conditional generation benchmark is introduced using eluted ligand data with a strict split where no 9-mer is shared across train/test clusters. On this task, FlexFlow greatly improves a held-out DeepMHCII-based discriminator score (rate guidance 0.58; latent guidance 0.66), while highlighting a quality–diversity tradeoff (latent guidance can improve plausibility while worsening embedding-distance coverage metrics).
📜Paper: arxiv.org/abs/2606.10543
#ComputationalBiology #GenerativeModels #FlowMatching #DiffusionModels #ProteinDesign #DNADesign #PeptideDesign #MHC #MachineLearning #Bioinformatics
3
16
1,423

13/25 𝗣𝗔𝗠𝗙: 𝗣𝗿𝗶𝗼𝗿-𝗔𝘄𝗮𝗿𝗲 𝗠𝘂𝗹𝘁𝗶𝗺𝗼𝗱𝗮𝗹 𝗙𝘂𝘀𝗶𝗼𝗻 𝗳𝗼𝗿 𝗜𝗻𝗰𝗼𝗺𝗽𝗹𝗲𝘁𝗲 𝗧𝗶𝗺𝗲 𝗦𝗲𝗿𝗶𝗲𝘀 𝗗𝗮𝘁𝗮
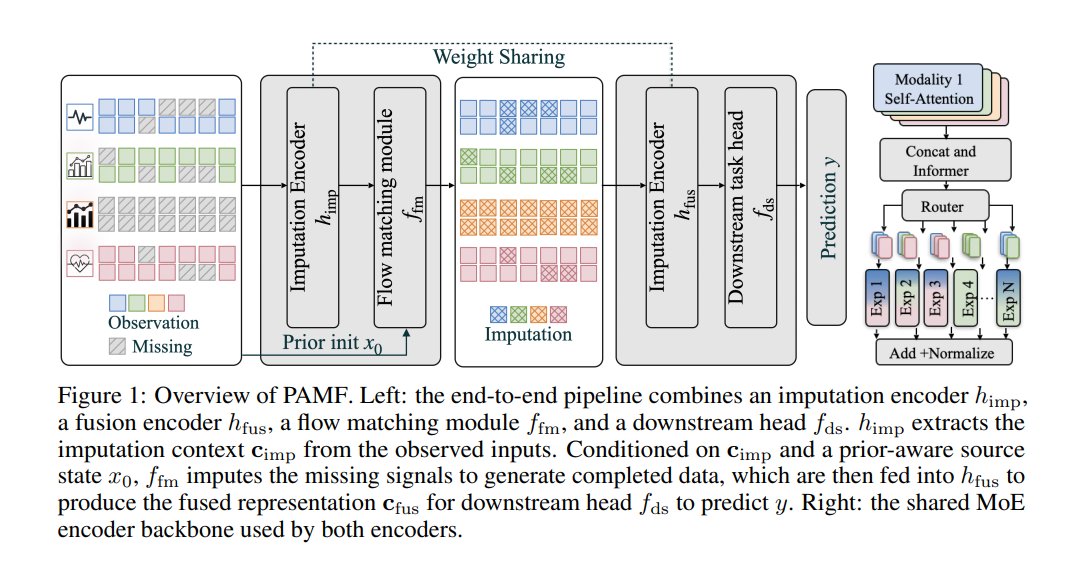
This paper presents PAMF, a multimodal time-series framework designed to address incomplete observations in healthcare data, specifically handling both within-modality and modality-level missing patterns. PAMF couples imputation with downstream prediction via prior-aware flow matching initialized with type-specific priors and architecturally matched encoders with weight sharing. Evaluated on multiple multimodal healthcare time-series benchmarks, PAMF achieves the strongest overall downstream performance across diverse datasets and missing settings compared to existing baselines.
#PAMF #MultimodalTimeSeries #MissingDataImputation #HealthcareAI #FlowMatching
Paper Link: arxiv.org/abs/2606.06328

1
13
Thanks to my coauthors Angela Lombardi, @MattAttimonelli , Giuseppe Fasano & @TommasoDiNoia at @sisinflab
#GenerativeAI #MedicalImaging #FlowMatching #Wavelets #Neuroimaging #MRI
45


Unsupervised source separation matches supervised models without clean training data. SURF uses flow matching with remixing bootstrap. Handles domain shifts. Demo released. #SourceSeparation #FlowMatching #MIR arxiv.org/abs/2606.04921v1
8


FlowMatchingふんわり理解。ベイズ的なモデル化はVAEと同じ:
x~N(0,1), φ(x;θ)~データ分布q
これをt=0の分布Nをt=1のqに変換した、と見て”時間発展演算子”φ_tにすると対応するベクトル場uが存在するからこれを学習。t=0,1の境界条件だけ決まってるからダイナミクスを設計するところに自由度がある
1
1
184

Geometric Flow Matching for Molecular Conformation Generation via Manifold Decomposition
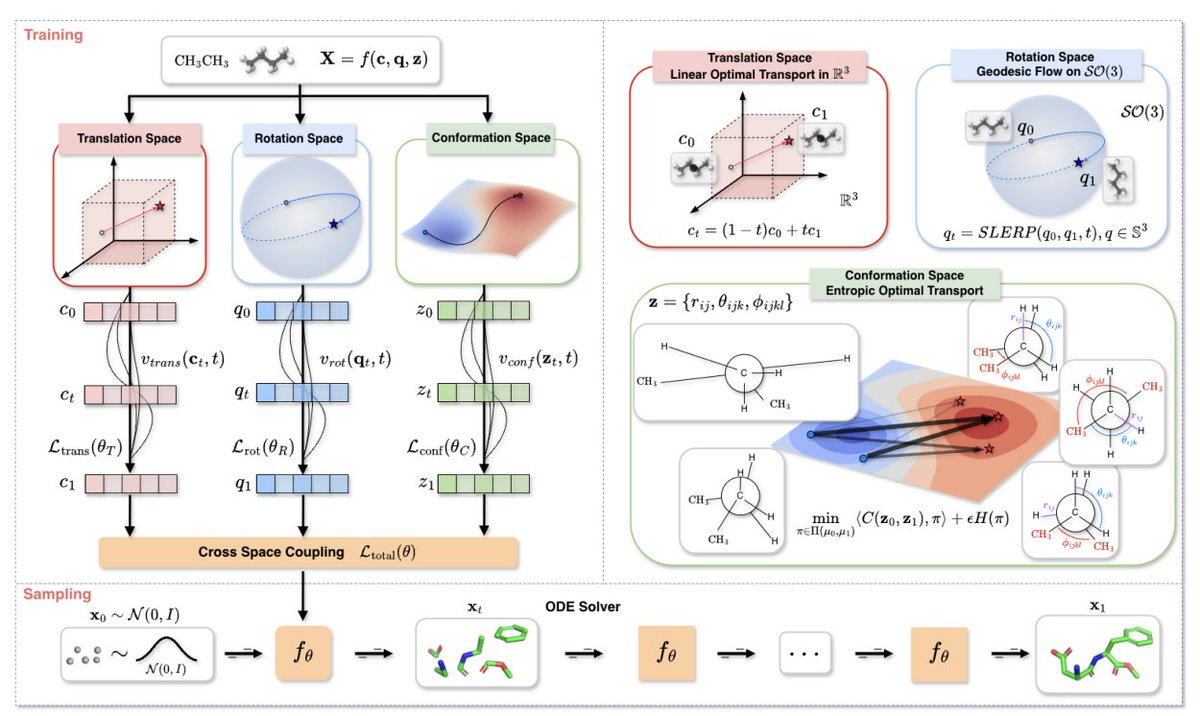
1. GO-Flow reframes molecular conformer generation as motion on the right manifolds rather than Cartesian point-cloud denoising, explicitly separating stiff vs. flexible degrees of freedom (rigid bond lengths/angles vs. flexible torsions) to avoid physically implausible intermediate structures.
2. The core idea is a manifold decomposition of a 3D molecule into three coupled subspaces: translation MT = R3 (center of mass), rotation MR = SO(3) (global orientation), and conformation MC (internal coordinates: bond lengths in R , bond angles in (0, π), torsions on a torus).
3. Each subspace uses a geometry-matched flow: translation uses linear optimal transport with constant velocity; rotation uses geodesic flows on SO(3) via unit quaternions and SLERP (avoiding Euler-angle singularities); conformation uses entropic optimal transport to traverse a multimodal internal-coordinate landscape without cutting through high-energy regions.
4. For rotation, GO-Flow trains on Lie-algebra angular velocities derived from relative quaternions (via log map), ensuring the learned velocity corresponds to a valid axis-angle rotation and remains consistent with rotation-equivariant neural architectures.
5. For internal coordinates, GO-Flow computes an entropic OT coupling (Sinkhorn) between noise and data internal-coordinate distributions, then defines the target velocity by barycentric projection of the OT plan; this is intended to better preserve diverse conformational modes while maintaining chemical plausibility.
6. The method composes the three learned velocities back into Cartesian atomic velocities using a Jacobian-based projection from internal coordinates to Cartesian space, yielding a total update that adds: translation, rotational cross-product motion around the center of mass, and J(z) vconf for internal-coordinate-driven motion.
7. Training uses a stability-oriented curriculum: (i) learn each manifold flow separately, (ii) jointly couple them with a Cartesian flow-consistency loss and uncertainty-based loss weighting, and (iii) optionally fine-tune as a continuous normalizing flow with likelihood evaluation using a Hutchinson trace estimator.
8. On GEOM-Drugs and GEOM-QM9, GO-Flow reports state-of-the-art coverage/matching tradeoffs while sampling with only 50 ODE solver steps; on GEOM-Drugs it achieves COV-R 94.82% and MAT-R 0.797 Å at 50 steps, compared with GeoDiff needing 5,000 steps for lower recall and worse matching.
9. Ablations indicate the conformation manifold (entropic OT over internal coordinates) contributes the largest gains; removing it causes the biggest drop in both recall and matching, while removing SO(3) rotation handling also noticeably degrades performance, supporting the claim that respecting manifold structure improves both validity and efficiency.
📜Paper: arxiv.org/abs/2605.25577
#ComputationalChemistry #MolecularGeneration #DiffusionModels #FlowMatching #OptimalTransport #SO3 #EquivariantML #DrugDiscovery #GeometricDeepLearning #ConformerGeneration
5
26
1,328


7/7 Paper project page code:
abhi-rf.github.io/egoflow/
arxiv.org/abs/2604.01421
github.com/abhi-rf/egoflow
Huge thanks to the great team of Huajian Zeng, @12Zuo, Daniel Cremers, and @xiwang1212
@tumcvg
#CVPR2026 #ComputerVision #EgocentricVision #EmbodiedAI #FlowMatching
1
1
220
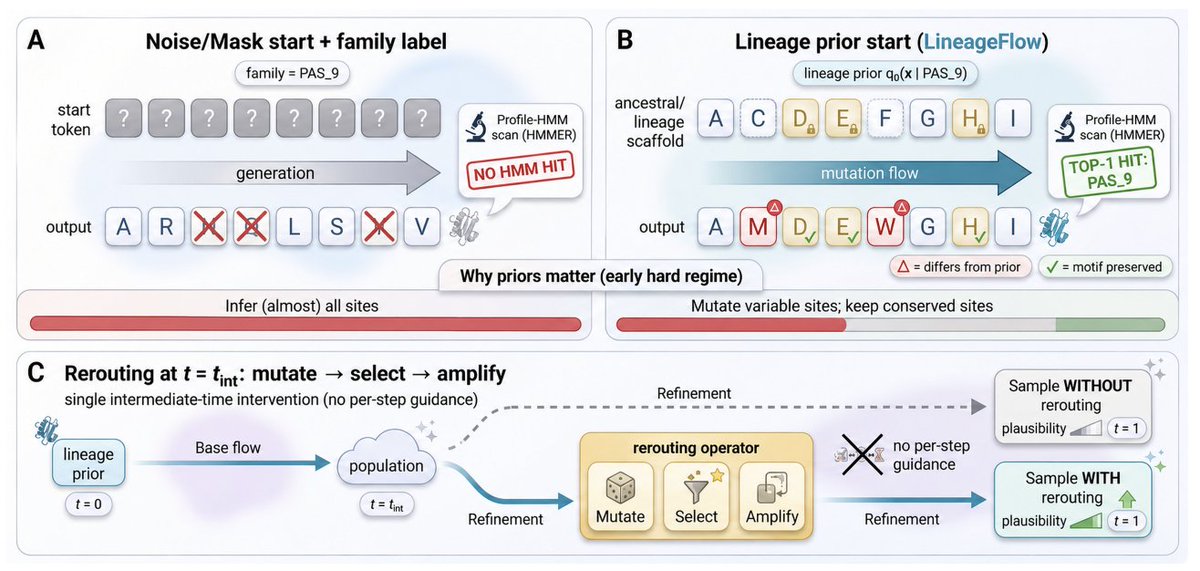
LineageFlow: Flow Matching for High-Fidelity Family-Aware Protein Sequence Generation
1. The paper argues that a key bottleneck in family-conditioned protein generation is the initialization prior: uniform-simplex noise or mask corruption erases evolutionary structure, forcing models to reconstruct conserved motifs “from scratch,” which weakens family control and plausibility.
2. LineageFlow replaces generic priors with lineage priors derived from ancestral sequence reconstruction (ASR): for each Pfam family, it infers a phylogeny from the MSA, performs marginal ASR at the root, and converts the site-wise root posterior into Dirichlet parameters used as a family-specific prior over the probability simplex.
3. With this design, generation is reframed as structured mutation from an evolved scaffold: conserved positions start concentrated, while variable sites retain uncertainty, aligning the trajectory with a family-specific manifold without feeding family labels or MSA prompts into the denoiser.
4. Methodologically, it builds on Dirichlet Flow Matching (DFM) on the simplex: each site follows an analytic Dirichlet path Dir(α(h,l) (tmax t) ei), with a derived lineage-specific vector field that conserves probability mass and keeps trajectories on the simplex.
5. Training uses a classifier parameterization: a transformer denoiser (initialized from ESM2) predicts terminal residues given (Xt, t), optimized by cross-entropy on valid (non-gap) MSA positions; the drift field is reconstructed by mixing analytic per-residue fields weighted by the predicted terminal distribution.
6. A second contribution is rerouting: a single intermediate-time inference intervention inspired by directed evolution (mutate → select → amplify) that steers samples toward a fitness objective without per-step gradient guidance, formalized as KL-regularized exponential tilting of the intermediate distribution.
7. Large-scale evaluation trains one shared model across 8,886 Pfam families (~8.94M sequences; 5% held-out per family) and scores generation by profile-HMM family validity (HMMER), foldability proxy (OmegaFold pLDDT), self-consistency (ESM-IF perplexity), novelty (MMseqs2 NN identity), and diversity (MMseqs2 clustering).
8. Results emphasize the role of priors: uniform-/mask-initialized baselines (DFM, EvoDiff) show essentially zero Pfam top-1 family accuracy under this strict HMM library scan, even when given explicit family labels; ASR prior alone (iid sampling) already yields high family validity, indicating ASR carries strong family signal.
9. LineageFlow with rerouting achieves near-natural family validity (Accfam 95.3% vs 96.6% for held-out natural sequences), improves foldability over prior-only and over several baselines (mean pLDDT 76.6), while keeping substantial novelty among foldable samples (Novelty@0.8 86.2%, Novelty@0.6 48.9%) and strong diversity.
10. A mechanistic analysis attributes gains to the “hard regime” at early times: Bayes-oracle denoising accuracy is higher under ASR priors than uniform priors when states are most corrupted, raising the recoverable signal ceiling and reducing early errors that propagate through the flow.
11. In a zero-shot enzyme case study, the denoiser is trained without three enzyme families, but priors are still built from their MSAs/trees; sampling without fine-tuning preserves motifs and novelty, and rerouting (using an unsupervised ESM2 plausibility objective) increases motif agreement and improves solubility/thermostability proxy distributions.
12. Limitations noted: reliance on high-quality MSAs and phylogenetic inference for priors; generation is tied to family alignment coordinates and does not model indels explicitly; evaluation relies on computational proxies (pLDDT, predictor-based properties) without experimental validation; rerouting adds compute and depends on the fitness function.
💻Code: github.com/Jinx-byebye/Linea…
📜Paper: arxiv.org/abs/2605.22252
#ComputationalBiology #ProteinDesign #GenerativeModels #FlowMatching #DiffusionModels #Phylogenetics #AncestralSequenceReconstruction #MachineLearning #Bioinformatics
7
32
2,856

May 24
Excited to share that our paper “Closed-Form Concept Erasure via Double Projections” has been accepted to #CVPR2026!
As generative models become increasingly powerful, concept erasure is emerging as an important problem for trustworthy and controllable AI. However, many existing approaches rely on iterative optimization, which can unintentionally damage non-target concepts and introduce additional computational overhead.
In this work, we revisit concept erasure from a geometric perspective.
We propose a simple and efficient closed-form framework based on sequential double projections, enabling concept removal through direct geometric operations rather than iterative optimization. Our method is lightweight, interpretable, and better preserves surrounding semantic structures during erasure.
The framework is applicable to both diffusion-based and flow-matching generative models, including FLUX.
This work is part of our broader effort toward trustworthy generative AI, controllable foundation models, and safer generative systems.
Paper: arxiv.org/abs/2604.10032
#CVPR2026 #GenerativeAI #DiffusionModels #FlowMatching #ConceptErasure #TrustworthyAI #FoundationModels
1
4
933

May 23
Why doesn't openfasttokenizer training on VLM first doesn't benefit final performance in flowmatching stage
56

May 21
🚀 Does high-quality audio generation really require latent compression?
Excited to share our new paper🚀🚀🚀
WavFlow: Audio Generation in Waveform Space.
WavFlow removes the audio tokenizer / VAE bottleneck and performs flow matching directly in raw waveform space.
Instead of generating compressed latent tokens, WavFlow directly models waveform patches end-to-end, achieving competitive or better performance than latent-based methods on both Video-to-Audio and Text-to-Audio benchmarks.
Key ideas:
• waveform patchify
• x-prediction flow matching
• amplitude lifting for stable raw-space training
• large-scale 5M video-text-audio training pipeline
Paper: arxiv.org/abs/2605.18749
Demo: facebookresearch.github.io/W…
Code: github.com/facebookresearch/…
#AI #AudioGeneration #DiffusionModels #FlowMatching #MetaAI
Pop your headphones on 🎧 , turn on the sound📣, and listen to what our model can do!
1
236

1step consistency autoencoders seem to heavily point towards something like, "you can directly learn a latent space that functions as a kind of coordinate system for a high dim manifold", without the many-step/stochastic high frequency detail stuff baked in vs. a flowmatching AE
10
386

May 18
Supertonic: ONNX 런타임 기반의 초경량 온디바이스 다국어 TTS 시스템 (feat. Supertone AI)
(by 9bow님)
d.ptln.kr/10247
#ondevice #texttospeech #flowmatching #supertone #multilingualtts #onnxruntime #supertonic
28
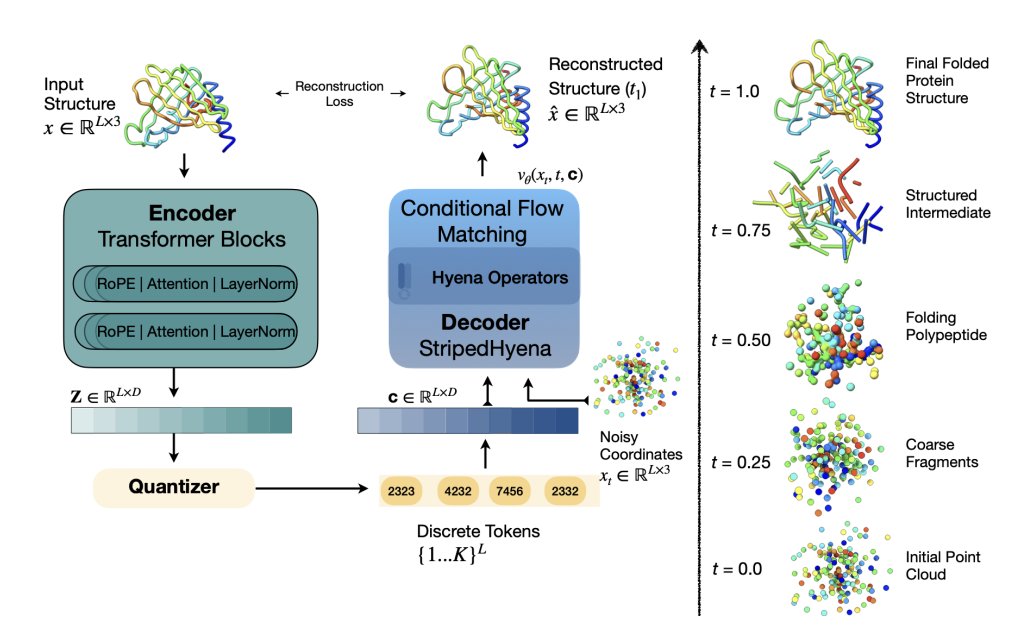
Yeti: A compact protein structure tokenizer for reconstruction and multi-modal generation
1 Yeti is a protein structure tokenizer designed not just for reconstruction, but explicitly to support multi-modal generation where sequence and 3D structure can be modeled together as discrete “language-like” tokens.
2 Core idea: convert continuous Cα coordinates (L x 3) into discrete tokens using Lookup-Free Quantization (LFQ), then reconstruct structures with a flow-matching decoder trained end-to-end (single-stage), rather than the common two-stage “train tokenizer then train bigger decoder” recipe.
3 The tokenizer uses an 8,192-entry codebook (K=8192) with latent dimension D=log2(K)=13; quantization is essentially a sign operation per dimension, making token lookup-free and simple to implement while still enabling a large vocabulary.
4 A key contribution is emphasizing that reconstruction quality and downstream generation quality are not the same objective; Yeti argues tokenizers should be evaluated on both reconstruction and token statistics (codebook utilization, diversity), because token collapse or repetition can hurt generative modeling.
5 On token statistics, Yeti reports consistently high entropy/perplexity and near-saturated intra-structure token diversity (about 98–99% unique-token fraction within a protein), suggesting tokens are information-dense and non-redundant across residues.
6 Reconstruction results show strong global topology recovery (TM-score ~0.95–0.97 across CAMEO/CASP14/CASP15/CATH for 50<L<=256). RMSD is competitive, with the model often ranking second-best while using far fewer parameters than large baselines (62.5M vs ~648M for ESM3).
7 Architecture choice matters: a StripedHyena decoder (hyena operators) trained more stably than a Transformer decoder in their experiments, maintaining better training dynamics and codebook utilization; this is one reason the tokenizer stays compact yet expressive.
8 For generation, they train a multi-modal masked diffusion model jointly over amino-acid sequence and Yeti structure tokens entirely from scratch (no pretrained initialization). The model (224M) performs unconditional co-generation of both sequence and structure in one unified process, instead of cascaded pipelines.
9 In co-generation evaluation (length bins up to 500 residues), the scratch-trained model achieves average self-consistency scTM ~0.70 and pLDDT ~76, with novelty vs CATH reported around 0.54 (lower similarity implies more novelty). ProteinMPNN recovers ~30.6% of the generated sequences from the generated structures, supporting designability.
10 The flow-matching reconstruction trajectory analysis suggests a two-phase behavior: early global compaction (radius of gyration decreases) followed by late “commitment” where secondary structure and global topology consolidate rapidly near the final steps (t > 0.9), offering a diagnostic lens into what the decoder learns.
📜Paper: arxiv.org/abs/2605.09981
#ProteinDesign #ProteinStructure #GenerativeAI #DiffusionModels #FlowMatching #MultimodalAI #ComputationalBiology #MachineLearning
7
49
9,025