
From @grok - happy hunting!
Here are the **most disqualifying** weaknesses, gaps, and holes that render the 2023 Cell ICE mice study (and associated Sinclair/OSK claims) largely **bunk** as proof of epigenetic information loss as a primary, causal, and safely reversible driver of mammalian aging:
- **No lifespan extension in the core ICE study** — The paper demonstrates accelerated aging phenotypes and partial molecular reversal but reports zero data on increased median or maximum lifespan in ICE mice after OSK treatment (or even in untreated controls). Claims of "reversing aging" collapse without this gold-standard endpoint.
- **Complete absence of teratoma, dysplasia, or cancer monitoring** — The word "teratoma" does not appear once in the paper despite well-documented oncogenic risks from in vivo OSK/OSKM reprogramming in prior studies; no systematic histology, tumor screening, or long-term safety data were provided.
- **Artificial induction does not mimic natural aging** — Repeated, targeted I-PpoI-induced DSBs in young mice create non-physiological, synchronized damage unlike the gradual, stochastic, diverse insults of normal aging; rDNA locus targeting introduces unruled-out confounds (rDNA instability affects lifespan in model organisms).
- **Whole-body, non-cell-autonomous design** — Systemic ICE activation prevents distinguishing direct cellular effects from secondary systemic responses; no tissue-specific controls.
- **Bulk-level data only; no single-cell resolution** — Epigenomic and transcriptomic analyses are bulk, masking cell-type heterogeneity, rare cell contributions, or variable reprogramming efficiency.
- **Mechanistic black box** — No identification of which specific chromatin factors relocalize, no in vivo chromatin contact mapping (Hi-C/HiChIP limited or absent for key claims), leaving the core "information loss via relocalization" hypothesis unproven at molecular detail.
- **Reversal is narrow, partial, and surrogate-heavy** — Phenotypic improvements limited to select tissues (retina, some kidney/muscle markers, neurons); relies heavily on epigenetic clocks and gene expression as proxies rather than comprehensive functional restoration across organs or organismal healthspan.
- **Ignores non-cellular aging components** — Extracellular matrix (e.g., irreversible collagen cross-links via glycation), persistent senescent cell effects, proteostasis decline beyond epigenetics, and other hallmarks remain unaddressed and unlikely fixed by transient OSK alone.
- **Overhyped translation to "age reversal"** — Media and author framing ("rebooting," "permanent reset," "aging forwards and backwards at will") far exceeds the data; independent replication with rigorous lifespan/safety endpoints is absent, and critics note the work leans more promotional than definitive.
These flaws mean the study provides interesting correlative or provocative data at best but fails as causal proof or a foundation for human "information theory of aging" therapies. Stronger, multifactorial models of aging (damage accumulation across compartments) remain intact. Independent, long-term studies with full organismal endpoints are essential before any claims graduate from hype.
2
2
750

Mar 24
Chiron3D, a DNA-only attention model initialized with Borzoi embeddings designed to predict CTCF HiChIP contact maps.
1
3
242
Mar 24
Chiron3D, a DNA-only attention model initialized with Borzoi embeddings designed to predict CTCF HiChIP contact maps.
1
2
552

29 Nov 2025
トウモロコシ近交系とF1雑種でHiChIP解析を行い、遺伝子型特異的な長距離クロマチン相互作用を同定。雑種では予期せぬ相互作用や対立遺伝子間相互作用が見られ、非相加的遺伝子発現や雑種強勢の仕組みを3D制御ネットワークから説明できる可能性が示された。Nat. Commun nature.com/articles/s41467-0…
2
14
741

4 Nov 2025
DiffHiChIP: Identifying differential chromatin contacts from HiChIP data dlvr.it/TP3LLJ
1
3
15
1,597

Teardown of the 2" LCD Screen Mirror device
~20€ From Aliexpress
s.click.aliexpress.com/e/_oC…
Surprisingly packed
- Unknown DH390D HT2522A SoC likely HiChip HC15xx 4MB SPI Flash
- Battery Powered
- Speaker
- Realtek WiFi Chip
- Jieli BLE SoC
Similar to youtu.be/pFBn6lMJ7q8




1
38
3,300

27 Aug 2025
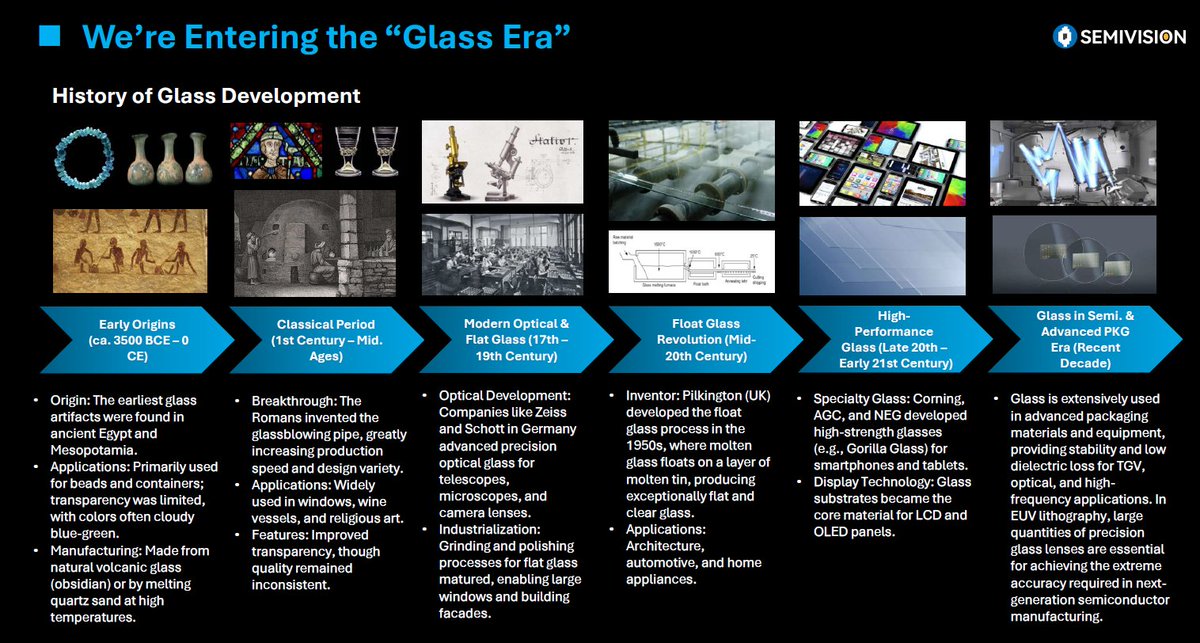
🥃#SemiVision: Today I had the honor of giving a talk at the #HICHIP Alliance, with the topic:“From Sand to Silicon — and Now to #Glass: The Evolution of Chip Packaging.”
This slide is something I spent a lot of time putting together. While preparing it, I also gained a much deeper appreciation for the long history of glass — from ancient artifacts to today’s role in advanced semiconductor applications.
Let’s walk through this journey together.
Hope you’ll enjoy it as much as I did!
10
561

12 May 2025
Our work on 3D genome organization in human cancer is out! In collaboration with @ZYanding @HowardYChang @WJGreenleaf and TCGA we used HiChIP to profile enhancer connectivity and rewiring in primary tumors.
nature.com/articles/s41588-0…
4
17
91
5,378

2 Mar 2025
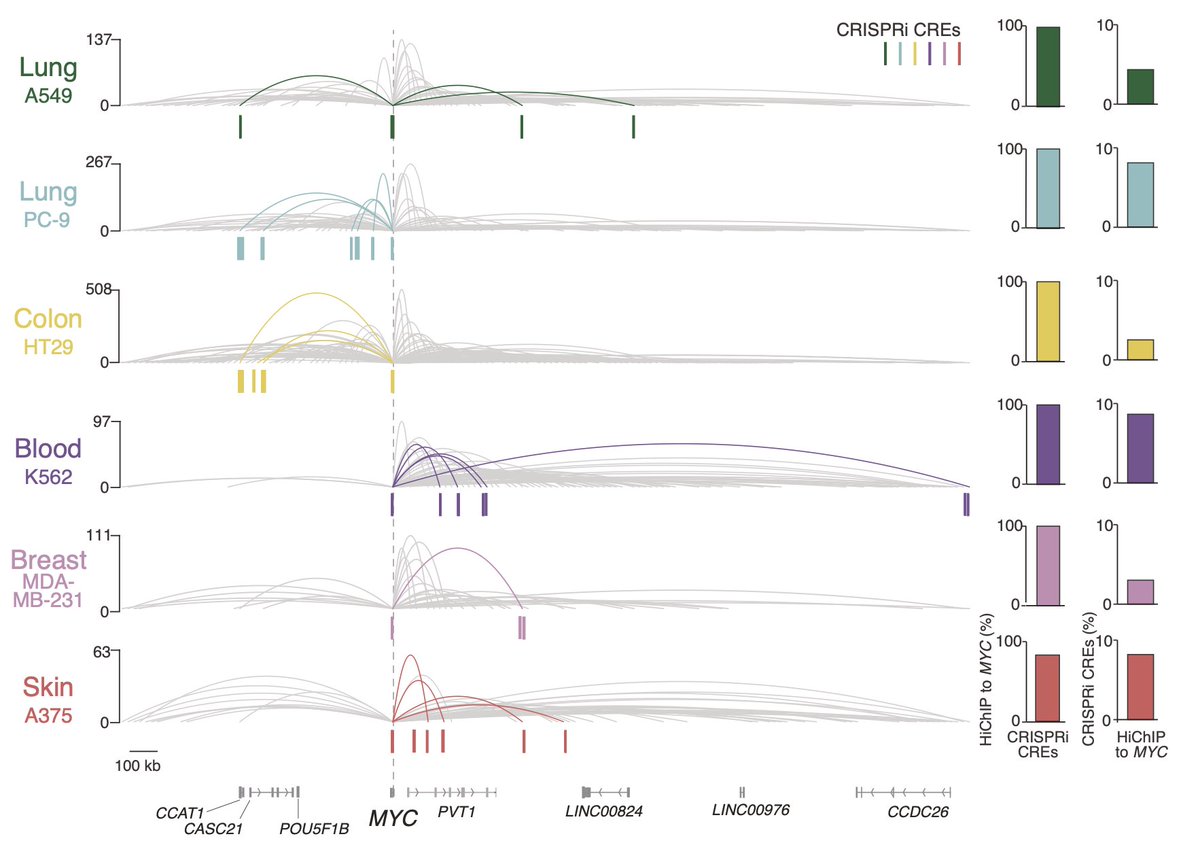
How are these CREs regulating MYC?
Using H3K27ac HiChIP we found that all but one of the CREs is in physical contact with the MYC promoter. ➰
However, contact 👉👈 is not enough for regulation: less than 10% of all contacts to the MYC promoter were identified as CREs.

1
3
340

27 Jan 2025
Please also check out two other papers using HiChIP and Hi-C to detect genetic associations with chromatin looping by Ferhat Ay's and Gisela Orozco's labs, which were submitted in parallel to ours and provide complementary insights:
nature.com/articles/s41467-0…
1
4
14
694

25 Jan 2025
Some updates. Chip is HiChip C3100, CPU Speed I think is actually 1.0GHz and 1.2G is some marketing thing, similar to RAM actually being 256 MegaBYTES which is 2 GigaBITS...I found a dump of the OS and some other useful stuff too
Also this pic of a partial teardown

1
1
44

20 Jan 2025
Extremely happy to share that our work DiffHiChIP: a method to identify differential chromatin interactions from HiChIP (and any other chromatin interaction data such as Hi-C, ChIA-PET), is now online.
Preprint in: biorxiv.org/content/10.1101/…
5
3
8
421

3 Jan 2025
Struggling to detect enhancer-promoter loops from Hi-C? Frustrated by stripes caused by biases in HiChIP data and unsure how to normalize it? Check out Raichu—a novel normalization method for chromatin contact data .biorxiv.org/content/10.1101/…
6
17
86
12,821
26 Dec 2024
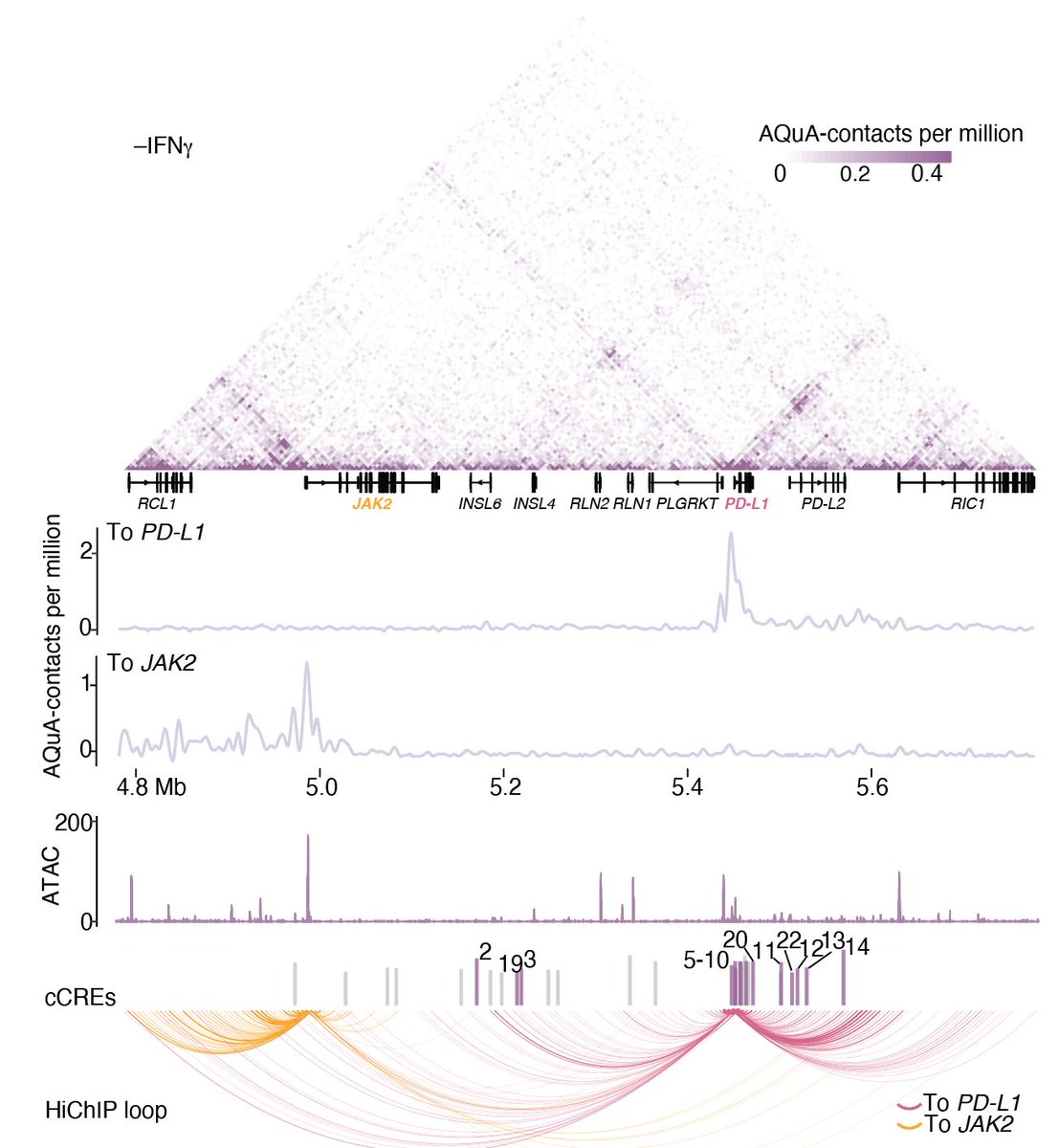
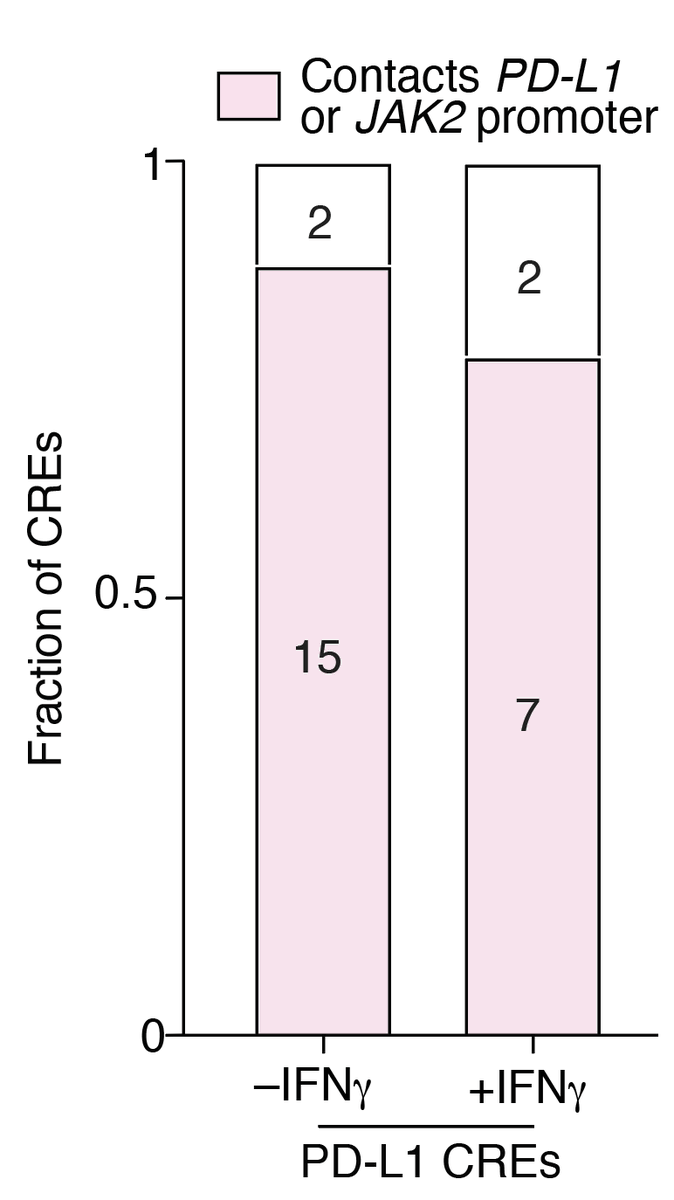
To test this, @XueXinhe & lab 3D genome guru 👩🔬#ChristinaCaragine performed enhancer HiChIP and found that many of the PD-L1 CREs that we validated individually contact the promoter of PD-L1 or the promoter of a nearby PD-L1 regulator (JAK2).
1
1
6
1,006

6 Dec 2024
Dovetail 使用論文情報
Dovetail社 HiChiP Kit を使用して多発性骨髄腫のスーパーエンハンサー制御遺伝子を明らかにした論文がNatureで発表されました。
This is a study utilizing Dovetail HiChIP and published in Nature: nature.com/articles/s41467-0…
12 Nov 2024

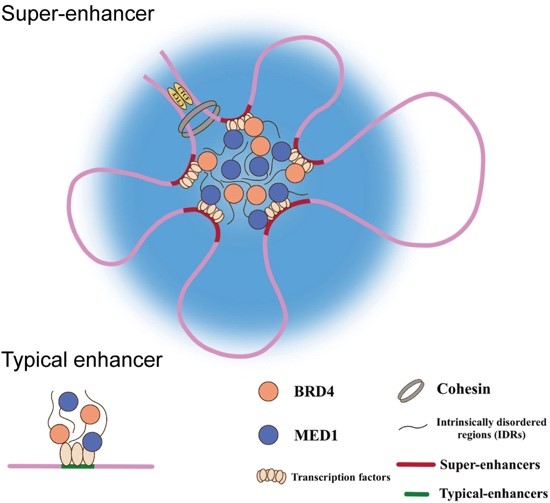
🧬 New Blog Alert: Targeting Super-Enhancer Driven Genes in Multiple Myeloma 🧬
👉 Read the full blog to learn more about these cutting-edge findings and their implications for cancer research here:
ow.ly/8lv050U5BPZ
#SuperEnhancers #DovetailGenomics #HiChIP
1
4
165

26 Aug 2024
This project was a joy, Ana knocked it out of the park with the HiChIP data. Chromatin looping by CTCF with and without DNA methylation!
25 Aug 2024

I am thrilled to see part of my postdoc work finally out! 🥳🥳. Thanks to @jrichardalbert and @maxvcg for the amazing work they have done and for being the best team one can imagine (together with the rest of the Greenberg lab) and of course, thanks to the rest of the authors!
6
336

15 Aug 2024
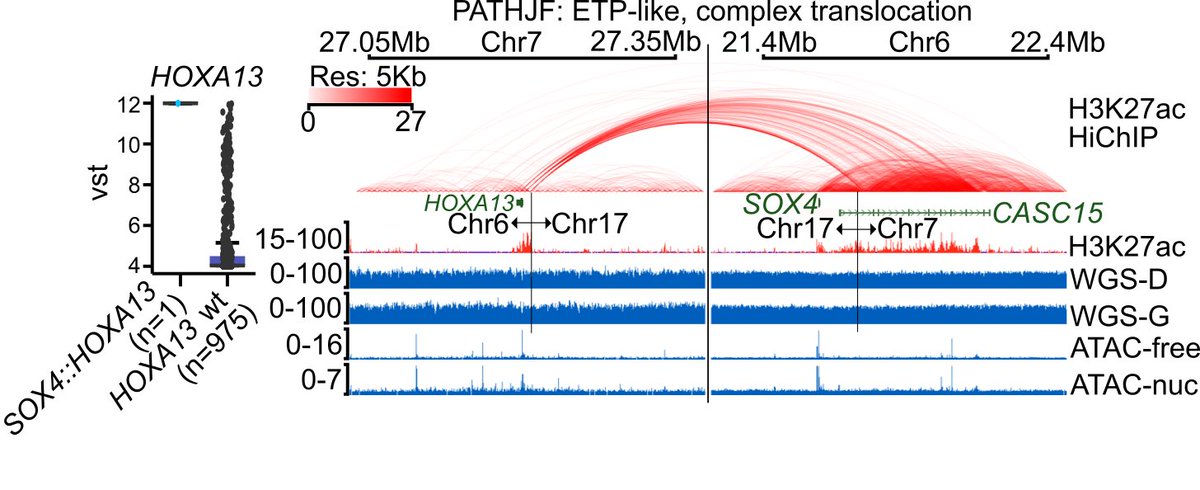
4) We conducted a targeted analysis of chromatin state and topology to elucidate the mechanistic consequences of enhancer perturbations, utilizing techniques such as HiChIP and ATAC-seq.
1
2
355

We used H3K27ac HiChIP to probe chromatin looping changes after 1 hour estrogen treatment. Thousands of loops change with a strong enrichment for estrogen receptor alpha (ER) bound regions. Genes with differential loops have larger transcriptional responses to estrogen 2/
1
62

30 May 2024
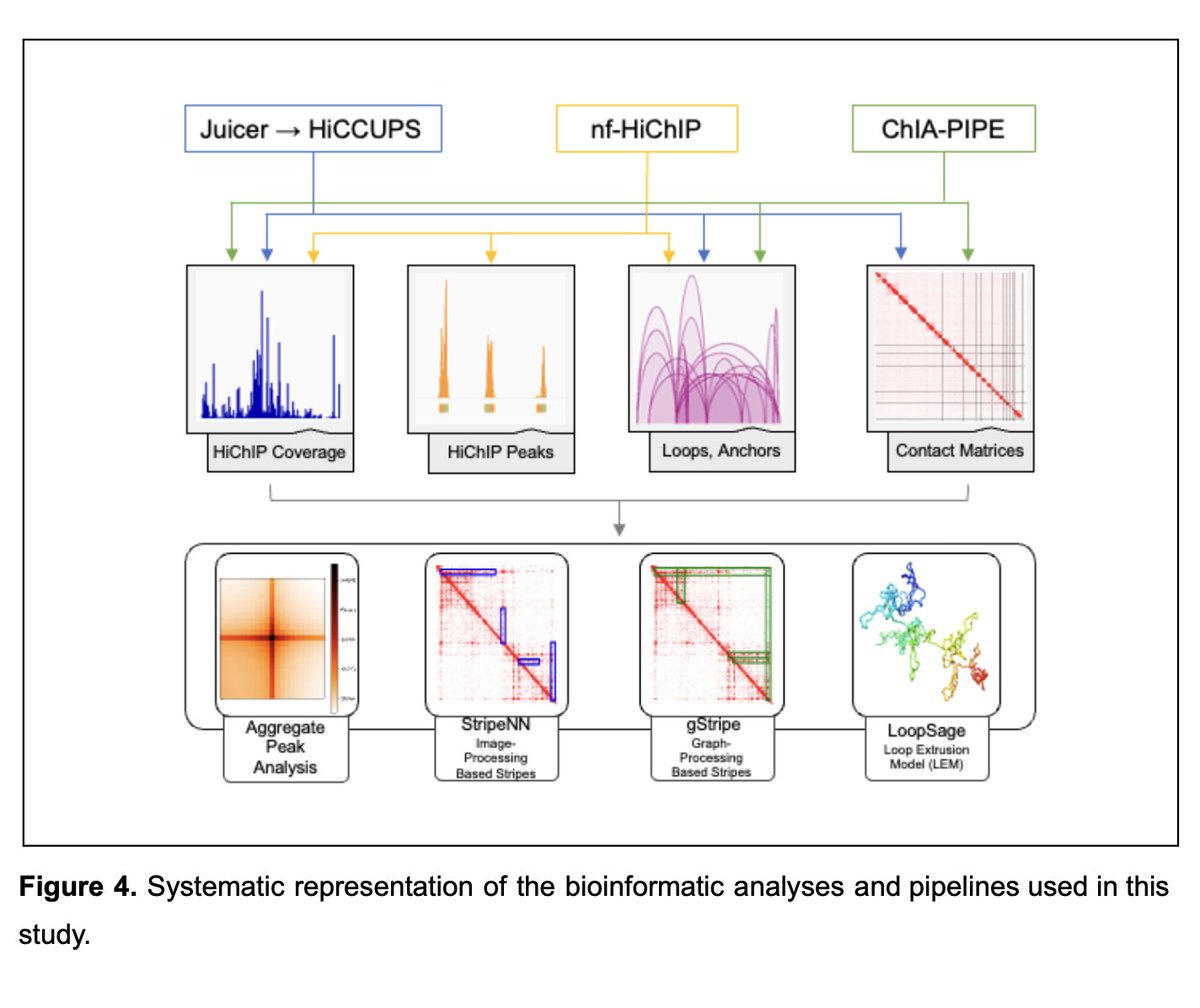
🛠 Improved cohesin HiChIP protocol and bioinformatic analysis for robust detection of chromatin loops and stripes
👏🏽 FA-EGS cross-linking HiChIP protocol improves stripe detection & enables building accurate loop extrusion models
bioRxiv (May 25, 2024) doi.org/10.1101/2024.05.16.5…
2
11
689

21 May 2024
Improved cohesin HiChIP protocol and bioinformatic analysis for robust detection of chromatin loops and stripes biorxiv.org/content/10.1101/… @4DNucleome
2
5
245