29 Jun 2025
Bimodal masked language modeling for RNA-seq and DNA methylation representation learning
1.This work introduces MOJO, a bimodal masked language model that learns joint representations of bulk RNA-seq and DNA methylation data using self-supervised learning. The model achieves state-of-the-art performance across cancer-type classification, survival prediction, and zero-shot clustering.
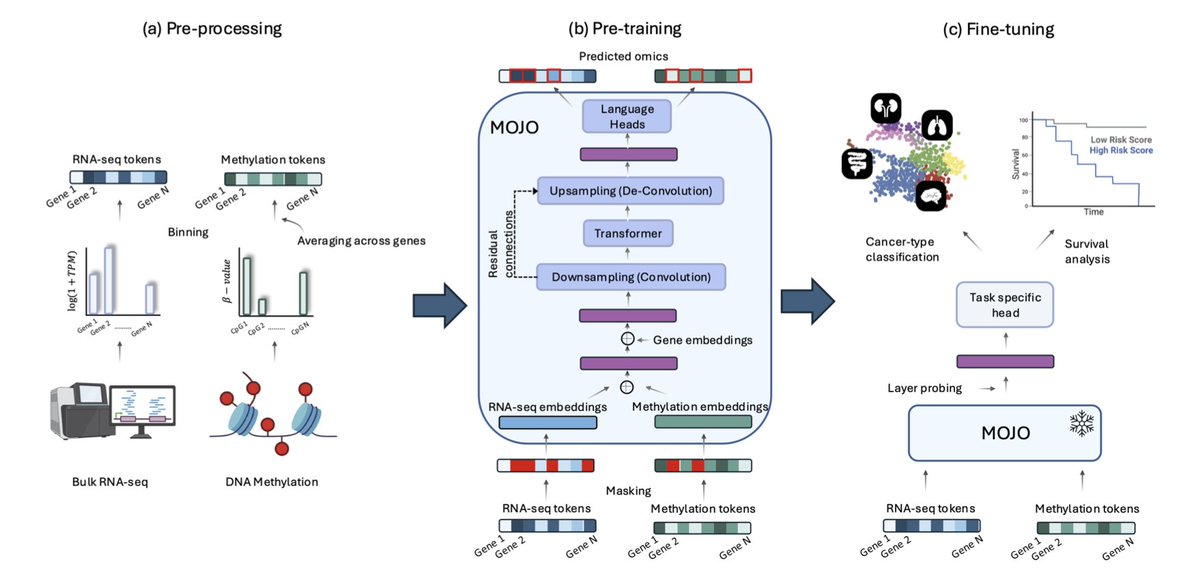
2.Unlike late integration approaches that combine separately pretrained models, MOJO is trained end-to-end on both modalities, capturing interactions between RNA-seq and methylation signals from the start. This joint modeling leads to improved generalization and representation quality.
3.MOJO combines convolutional layers with transformers to handle the long-range dependencies and high dimensionality of omics data efficiently. This hybrid architecture enables 100× faster training steps compared to purely transformer-based bimodal models.
4.The model is pre-trained on over 9,000 samples from the TCGA dataset using a bimodal masked language modeling objective, which corrupts 15% of tokens across both modalities to encourage joint representation learning.
5.In pan-cancer classification (33 cancer types), MOJO outperforms strong baselines including BulkRNABert, MethFormer, and CustOmics. It achieves a test macro-F1 of 0.935 and weighted-F1 of 0.952, showing notable gains even over sophisticated late integration methods using cross-attention.
6.For survival analysis, MOJO again outperforms unimodal and late integration baselines, achieving a C-index of 0.771 and weighted C-index of 0.670. These results indicate robust modeling of time-to-event prediction using joint omics embeddings.
7.The learned embeddings show strong zero-shot performance in breast cancer subtyping (PAM50) and pan-cancer cohort clustering, with MOJO achieving 0.777 accuracy for PAM50 and 0.928 for pan-cancer—better than late integration.
8.To tackle the real-world challenge of missing modalities, the authors propose two mechanisms: (i) re-training with incomplete modality samples (MOJO-MMO), and (ii) a mutual information auxiliary loss during fine-tuning, which regularizes the model to produce consistent outputs across partial inputs.
9.When either RNA-seq or methylation is missing at test time, the mutual information strategy allows MOJO to recover performance close to modality-specific models (e.g., from 0.538 to 0.916 weighted-F1 when RNA-seq is absent).
10.Overall, MOJO offers a scalable, performant, and robust approach to multi-omics integration, making it well-suited for clinical applications involving heterogeneous and incomplete datasets.
💻Code: github.com/instadeepai/multi…
📜Paper: biorxiv.org/content/10.1101/…
#RNAseq #DNAmethylation #MultiOmics #MaskedLanguageModel #Cancer #RepresentationLearning #Bioinformatics #SelfSupervised #TCGA #SurvivalAnalysis
3
9
1,001
7 May 2025
CreoPep: A Universal Deep Learning Framework for Target-Specific Peptide Design and Optimization
1. CreoPep is a generative deep learning platform that enables target-specific peptide design by combining masked language modeling with progressive masking and energy-based screening—offering a scalable alternative to traditional mutagenesis approaches.
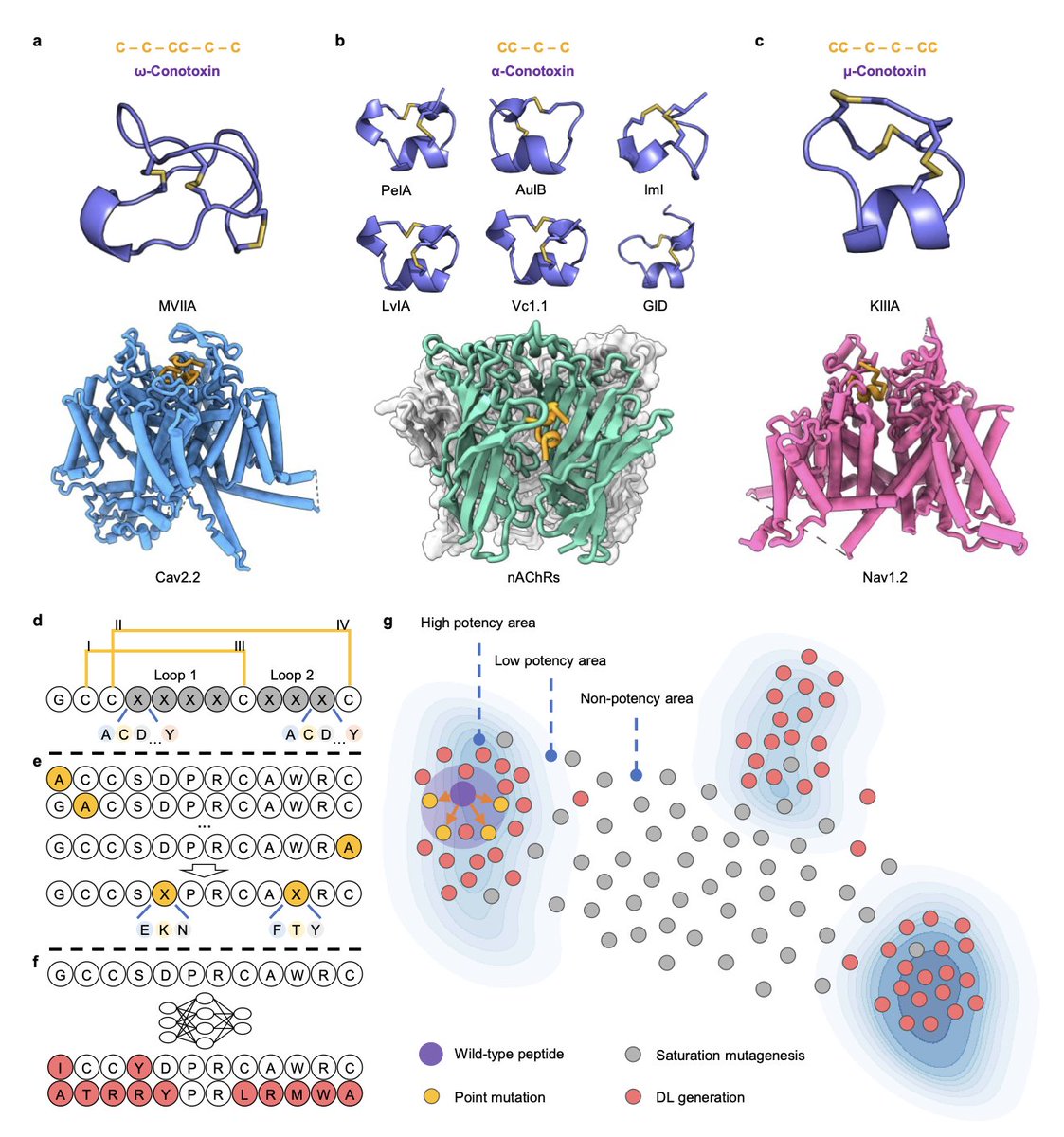
2. The framework focuses on conotoxins—peptides from cone snail venom known for their high affinity to ion channels like α7 nAChRs—and achieves submicromolar potency in designed variants, confirmed via electrophysiological assays.
3. CreoPep introduces a Progressive Masking (PM) strategy that gradually increases noise during training and denoises step-by-step during generation, improving contextual learning of sequence-function relationships beyond static masking schemes.
4. The model integrates multiple objectives—label prediction, conditional generation, and optimization—and uses subtype/potency annotations and auxiliary peptide inputs to guide design, allowing functional specificity without structural input.
5. CreoPep incorporates an energy-guided data augmentation pipeline powered by FoldX. Iterative ∆∆G screening of generated peptides filters high-affinity variants, producing an 8.8x expansion of the initial conotoxin dataset and enhancing diversity and performance.
6. With temperature-controlled multinomial sampling, CreoPep balances the generation of potent peptides with exploration of diverse sequences. Model outputs retain essential features like net charge, hydropathy, and disulfide patterns while accessing new chemical space.
7. Structural modeling via AlphaFold3 and FoldX confirms that top candidates like CP\_α7\_1 and CP\_α7\_6 bind the α7 nAChR with unique modes—one preserving canonical disulfide bridges, the other succeeding despite lacking them.
8. Electrophysiological validation on hα7 nAChRs showed that 7 out of 13 CreoPep-designed peptides achieved >50% inhibition at 10 μM, and two (CP\_α7\_1 and CP\_α7\_6) reached submicromolar IC50s (\~400–500 nM).
9. Sequence and structural analysis identified both conserved residues essential for activity (e.g. R7, W10, R11) and mutable positions that tolerate optimization, validating CreoPep’s capacity to extract functional design rules.
10. CreoPep offers a generalizable peptide engineering platform with potential applications across synthetic biology, drug discovery, and personalized therapeutics. Future directions include non-natural amino acid incorporation and multitarget design.
💻Code: github.com/gc-js/CreoPep
📜Paper: arxiv.org/abs/2505.02887
#PeptideDesign #DeepLearning #Conotoxin #Therapeutics #MaskedLanguageModel #DrugDiscovery #CreoPep #ProteinDesign #Bioinformatics #TargetSpecificDesign
2
14
1,012
7 May 2025
CreoPep: A Universal Deep Learning Framework for Target-Specific Peptide Design and Optimization
1. CreoPep is a generative deep learning platform that enables target-specific peptide design by combining masked language modeling with progressive masking and energy-based screening—offering a scalable alternative to traditional mutagenesis approaches.
2. The framework focuses on conotoxins—peptides from cone snail venom known for their high affinity to ion channels like α7 nAChRs—and achieves submicromolar potency in designed variants, confirmed via electrophysiological assays.
3. CreoPep introduces a Progressive Masking (PM) strategy that gradually increases noise during training and denoises step-by-step during generation, improving contextual learning of sequence-function relationships beyond static masking schemes.
4. The model integrates multiple objectives—label prediction, conditional generation, and optimization—and uses subtype/potency annotations and auxiliary peptide inputs to guide design, allowing functional specificity without structural input.
5. CreoPep incorporates an energy-guided data augmentation pipeline powered by FoldX. Iterative ∆∆G screening of generated peptides filters high-affinity variants, producing an 8.8x expansion of the initial conotoxin dataset and enhancing diversity and performance.
6. With temperature-controlled multinomial sampling, CreoPep balances the generation of potent peptides with exploration of diverse sequences. Model outputs retain essential features like net charge, hydropathy, and disulfide patterns while accessing new chemical space.
7. Structural modeling via AlphaFold3 and FoldX confirms that top candidates like CP\_α7\_1 and CP\_α7\_6 bind the α7 nAChR with unique modes—one preserving canonical disulfide bridges, the other succeeding despite lacking them.
8. Electrophysiological validation on hα7 nAChRs showed that 7 out of 13 CreoPep-designed peptides achieved >50% inhibition at 10 μM, and two (CP\_α7\_1 and CP\_α7\_6) reached submicromolar IC50s (\~400–500 nM).
9. Sequence and structural analysis identified both conserved residues essential for activity (e.g. R7, W10, R11) and mutable positions that tolerate optimization, validating CreoPep’s capacity to extract functional design rules.
10. CreoPep offers a generalizable peptide engineering platform with potential applications across synthetic biology, drug discovery, and personalized therapeutics. Future directions include non-natural amino acid incorporation and multitarget design.
💻Code: github.com/gc-js/CreoPep
📜Paper: arxiv.org/abs/2505.02887
#PeptideDesign #DeepLearning #Conotoxin #Therapeutics #MaskedLanguageModel #DrugDiscovery #CreoPep #ProteinDesign #Bioinformatics #TargetSpecificDesign
29
129
5,802
9 Apr 2025
Genomic Tokenizer: Toward a biology-driven tokenization in transformer models for DNA sequences
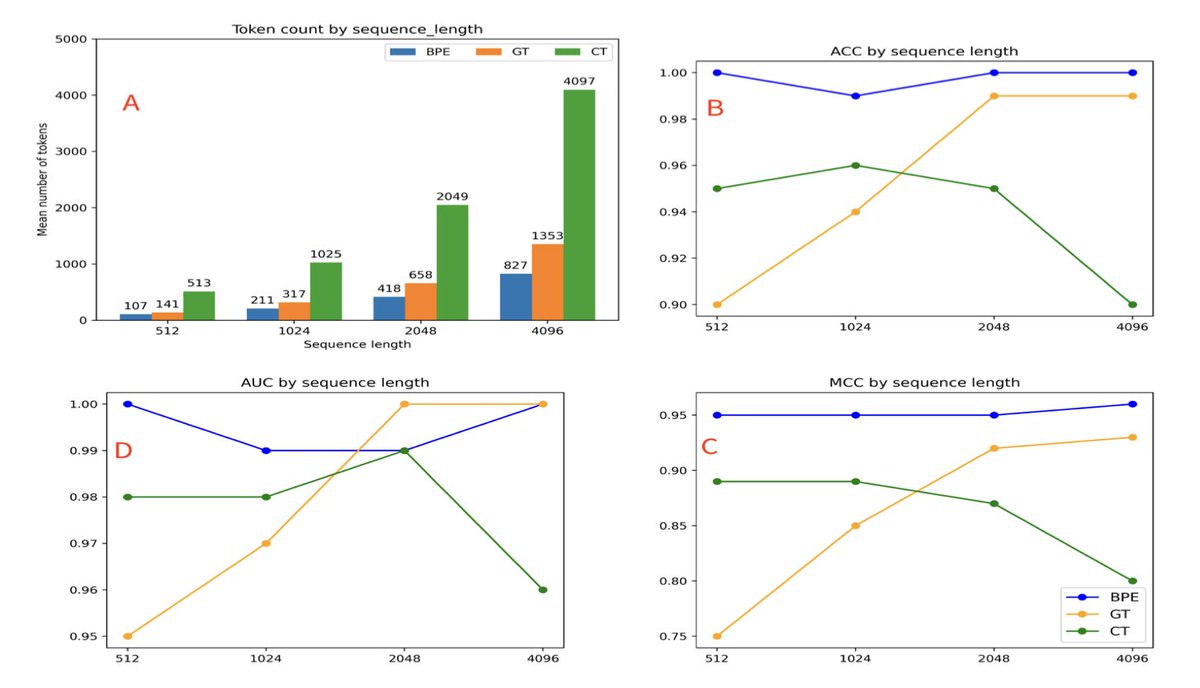
1. Genomic Tokenizer (GT) introduces a biologically grounded approach to DNA sequence tokenization, aligning with the central dogma of molecular biology by using codons—three-letter nucleotide sequences—as the core unit of tokenization.
2. Unlike traditional character or k-mer tokenizers, GT recognizes start and stop codons, assigns identical tokens to synonymous codons, and treats introns and out-of-frame regions as UNK tokens, reducing vocabulary size while preserving biological relevance.
3. GT is implemented within the HuggingFace tokenizer framework, enabling seamless integration into existing transformer-based DNA analysis pipelines and support for tasks like masked language modeling and sequence classification.
4. The tokenizer supports customizable start/stop codons and intron treatment, making it adaptable for different organisms, including prokaryotes and mitochondrial genomes.
5. In classification experiments using a lung cancer-related variant dataset, GT showed greater robustness to long sequence lengths compared to character tokenization, and outperformed it in longer sequence tasks.
6. While byte-pair encoding (BPE) achieved the highest overall performance, its large vocabulary comes with high computational cost. GT balances biological insight with computational efficiency and a compact vocabulary.
7. GT tokenization avoids issues of redundancy and information leakage in masked language modeling that are common in overlapping k-mer tokenizers, leading to cleaner training signals and potentially better generalization.
8. The biological foundation of GT allows it to better model frame-shift mutations, synonymous substitutions, and stop-gain variations—key features in predicting phenotypic impact from genetic data.
9. Preliminary comparisons highlight GT’s strength in biological modeling and suggest potential advantages for foundational model training across genomics tasks when compared with purely data-driven tokenizers.
10. GT is open-source, installable via PyPI, and encourages broader exploration across genomic datasets and transformer architectures, including long-context models such as HyenaDNA.
💻Code: github.com/dermatologist/gen…
📜Paper: biorxiv.org/content/10.1101/…
#Genomics #Tokenization #Transformers #Bioinformatics #DNASequence #LLM #Codon #MaskedLanguageModel #DeepLearning #HuggingFace
5
18
1,383