May 31
Claude also compared several tools for building the protein from the cryo-EM density. In this case, Claude used ModelAngelo, CryoAtom, and Cryo2Struct, and then constructed this dashboard to display the results.
1
5
732
Modeling Atomic Conformational Ensembles of Proteins via Test-Time Supervision of Boltz-2 on Cryo-EM Density Maps
1. The paper proposes a unified alternative to the standard two-stage pipeline (build atomic models from cryo-EM maps, then train ensemble predictors): it fine-tunes a pre-trained structure model (Boltz-2) directly on raw cryo-EM density map ensembles, so supervision happens in map space rather than requiring ground-truth atomic ensembles.
2. The resulting method, CryoSampler, targets “atomic ensemble model building”: given an ensemble of heterogeneous cryo-EM volumes from a single experiment (multiple conformational states), it outputs a matching ensemble of atomistic conformations with strong map–model agreement and chemically plausible geometry.
3. Key design choice: Boltz-2 is used as a frozen static trunk to produce a reference structure Xref, and CryoSampler learns deformations (per-atom coordinate offsets) on top of that reference to explain the cryo-EM density variability.
4. Training stage 1 is a 3D spatial VAE that encodes each cryo-EM map into a latent grid and decodes it into a 3D feature grid; a lightweight per-atom MLP queries this grid at the Boltz-2 reference coordinates to predict atomic offsets, producing an atomic model that is differentiably rendered back into a density map via a cryo-EM forward model.
5. Supervision is end-to-end through a volumetric reconstruction objective: the main signal is 1 − NCC between predicted and observed maps (map–map agreement), which avoids needing experimentally curated atomic conformations for each heterogeneous cryo-EM state.
6. To prevent “fitting the map at all costs,” CryoSampler adds structural regularization: differentiable MolProbity-style terms (clashes, Ramachandran, rotamers) plus a backbone soft rigid-body constraint that preserves local secondary-structure geometry relative to the Boltz-2 reference.
7. Training stage 2 freezes the VAE and learns a latent diffusion model using flow matching on the VAE latent codes; this provides a generative prior over the learned latent space, enabling sampling of multiple conformations rather than only reconstructing given maps.
8. Model-building results on four systems (TRPV3, integrin αVβ8, neurokinin-1 GPCR, human P-glycoprotein) show higher map–model fit than baselines (Boltz-2 alone, E2GMM, ModelAngelo, CryoBoltz where feasible), while maintaining competitive stereochemistry; the paper notes an explicit tradeoff where removing structural losses increases correlation but yields severely invalid geometry.
9. A practical systems point: some inference-time cryo-EM-guided baselines can fail on large complexes (e.g., CryoBoltz OOM on TRPV3 at 2,556 residues), while CryoSampler’s training-time adaptation is presented as a route to accurate fitting with controlled stereochemistry.
10. Beyond per-experiment overfitting, the paper reports preliminary in-domain generalization within TRP channels: after training on TRPV3 map states, CryoSampler samples an ensemble for TRPV5 without using TRPV5 cryo-EM at inference, and matches held-out TRPV5 maps better than Boltz-2 sampling or normal mode analysis (evaluated via ensemble-level precision/recall and Wasserstein distance derived from CCvolume).
📜Paper: arxiv.org/abs/2605.09832
#CryoEM #ProteinDynamics #DiffusionModels #ProteinStructure #Boltz2 #GenerativeAI #ComputationalBiology #StructuralBiology #ModelBuilding #EnsembleModeling

1
4
22
1,896

Feb 15
Very excited to share our work, a fish herpesvirus' cryo EM structure. It features a long PVAT tail, with new and host-like proteins discovered using modelAngelo, Dali, and Viro3D, including a host-like macrodomain
doi.org/10.64898/2026.02.13.…
1
9
505

27 Oct 2025
Accurate protein structure determination from cryo-EM maps using deep learning and structure prediction
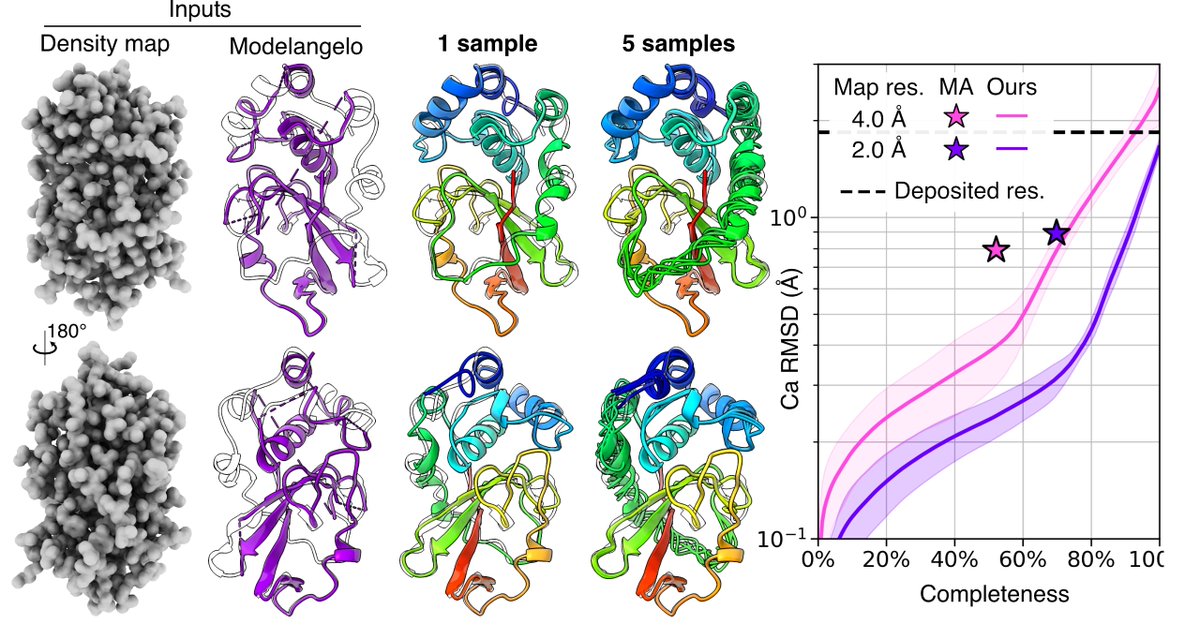
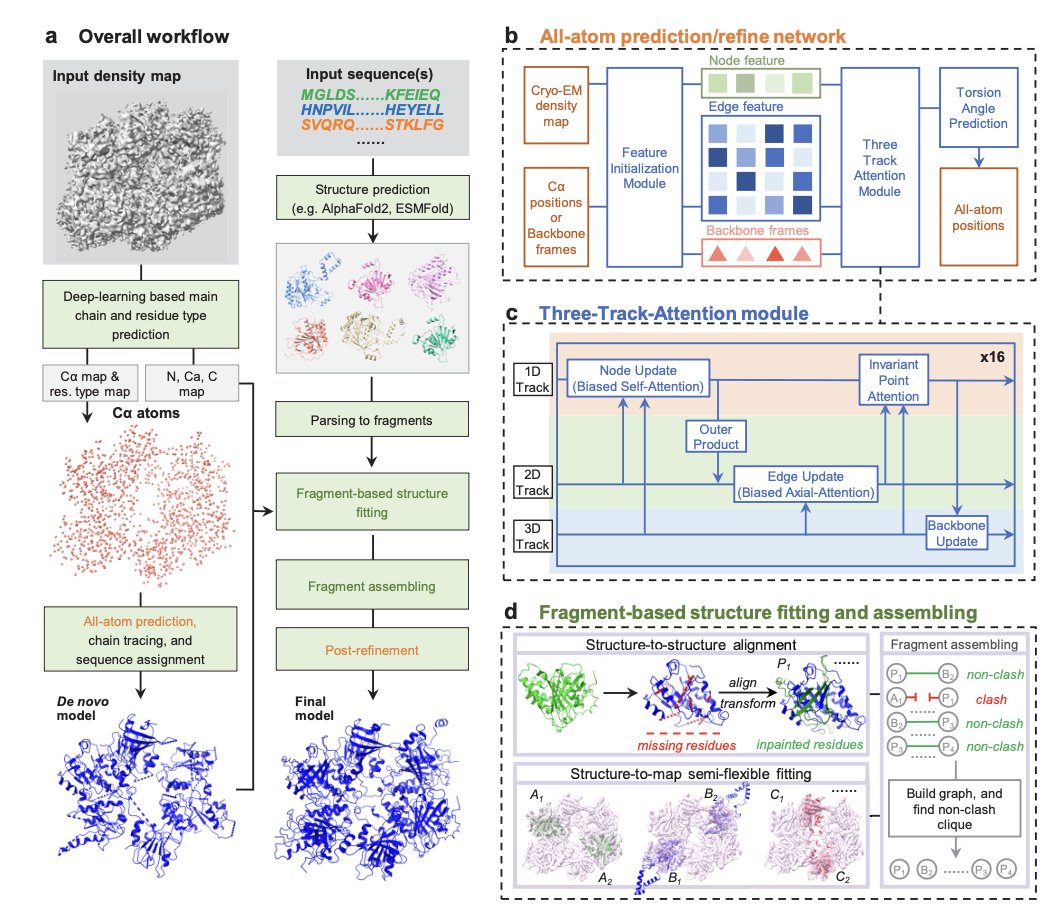
1. A new method named EMProt is introduced, which integrates deep learning and structure prediction to accurately determine protein structures from cryo-EM maps. This method is particularly innovative for its ability to handle the challenges posed by the intrinsic resolution heterogeneity of cryo-EM maps.
2. EMProt uses a three-track attention network to integrate map information and structure prediction. It first predicts amino acid types and Cα atom positions from raw density maps, then builds the all-atom structure by integrating 1D and 2D density features with 3D backbone frames.
3. The method has been extensively tested on a diverse set of 177 experimental cryo-EM maps with resolutions up to <4.0 ̊A. EMProt significantly outperforms existing methods like DeepMainmast, ModelAngelo, phenix.dock and rebuild, and AlphaFold3 in recovering the protein structure and building the complete structure.
4. EMProt’s ability to integrate predicted structures through a combination of structure alignment and FFT-based structure-to-map fitting is a key innovation. This allows it to refine and correct the built models, improving both model-to-map fit and overall accuracy.
5. The study also highlights EMProt’s performance in model-to-map fit validation and structure validation, showing high accuracy in terms of geometric constraints and model consistency. This suggests that EMProt can produce reliable models ready for deposition into databases like the PDB.
6. EMProt is freely available for academic or non-commercial users, making it a valuable tool for researchers in structural biology and related fields. The method’s effectiveness in integrating experimental data with predicted structures sets a new standard for cryo-EM structure determination.
📜Paper: biorxiv.org/content/10.1101/…
#CryoEM #DeepLearning #ProteinStructure #StructuralBiology #Bioinformatics
4
19
1,762

15 Oct 2025
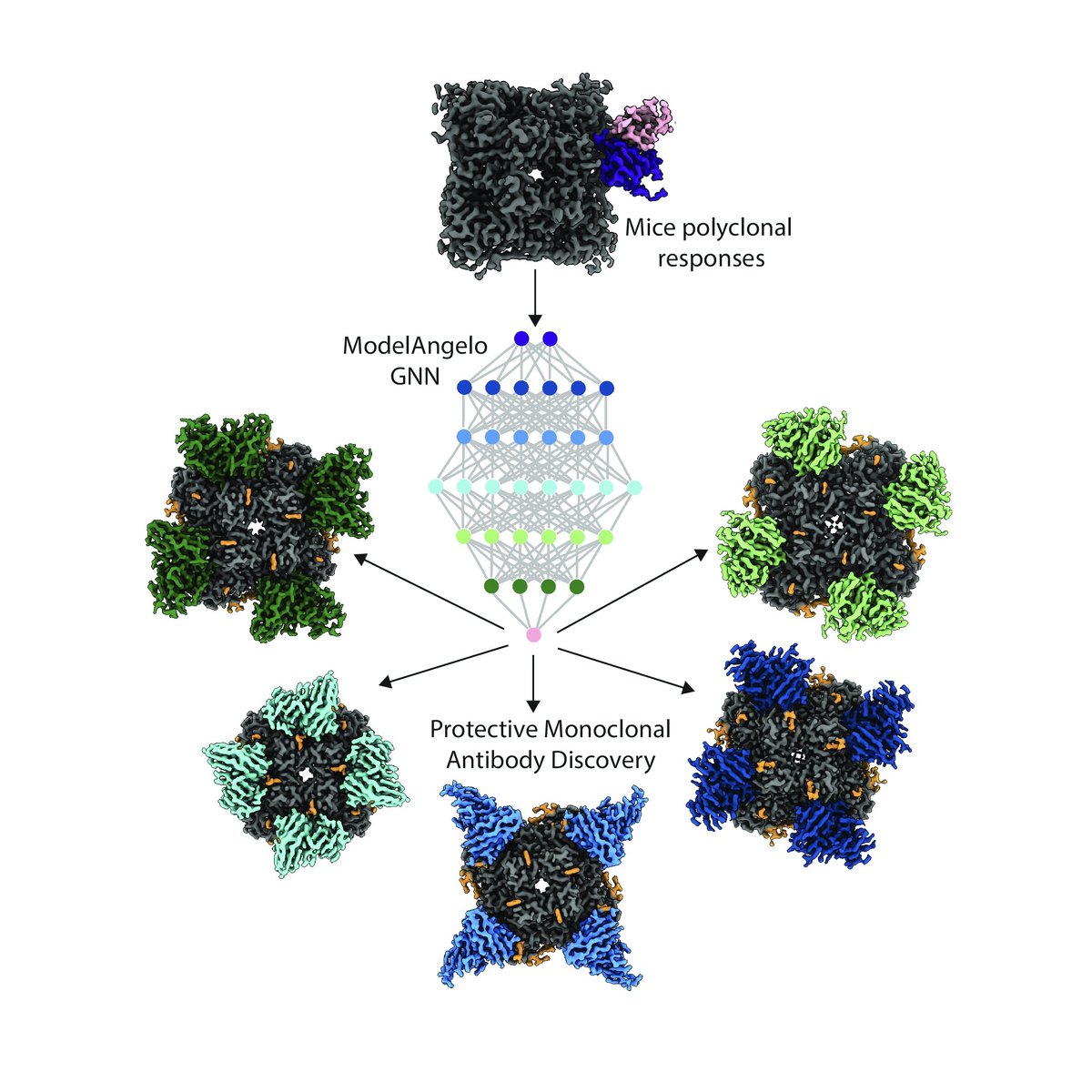
In a new @ScienceAdvances study, Andrew Ward (@WardLab1) and James Ferguson highlight a method that uses cryoEM and the AI tool ModelAngelo to rapidly identify the most protective monoclonal antibodies, cutting discovery time from weeks to under a day and accelerating pandemic preparedness. More: ow.ly/ifMg50XbWrJ
1
3
2
1,775

18 Sep 2025
🚀 Accelerated antibody discovery: Cryo-EM ModelAngelo to decode polyclonal antibody sequences in <1 day—boosting speed & precision 10x over traditional methods. 💉🔬 #Immunology #CryoEM #AntibodyDiscovery #AIinBiotech science.org/doi/10.1126/scia…
1
6
535
16 Sep 2025
In a new @ScienceAdvances study, Scripps Research Prof. Andrew Ward (@WardLab1) and staff scientist James Ferguson describe a method that combines cryo-electron microscopy with the graphical neural network-based structure building tool, ModelAngelo, to identify monoclonal antibodies from polyclonal responses.
This Structure-to-Sequence (STS) approach reduces the time needed to identify protective antibodies from weeks to under a day, offering a scalable method that minimizes data bottlenecks and accelerates therapeutic antibody discovery—critical for pandemic preparedness.
More at: ow.ly/JLUv50WW1wU

7
16
2,005
29 Jul 2025
Review of Deep Learning Applications to Structural Proteomics Enabled by Cryogenic Electron Microscopy and Tomography
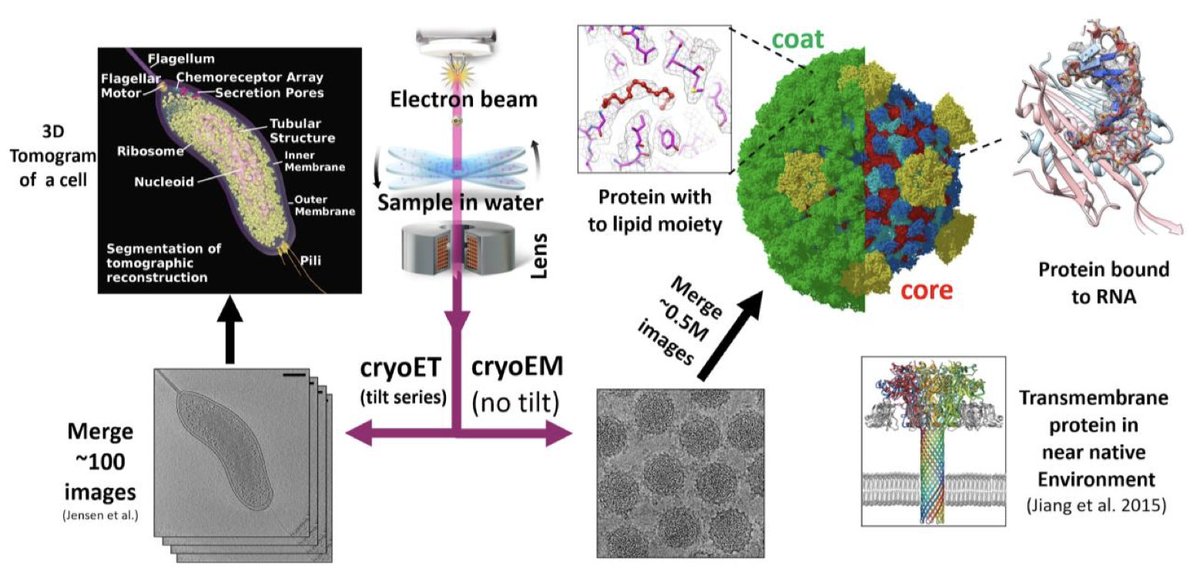
1. The integration of deep learning into cryoEM and cryoET workflows has revolutionized structural biology by addressing long-standing challenges such as low signal-to-noise ratios, preferred orientation artifacts, and missing-wedge problems. These advancements have led to remarkable achievements including near-atomic resolution reconstructions with minimal manual intervention.
2. Automated particle picking using convolutional neural networks (CNNs) like Topaz, crYOLO, and CryoSegNet has significantly improved efficiency and accuracy. CryoSegNet, in particular, combines a U-Net with a pre-trained foundation model to achieve state-of-the-art performance in particle detection and segmentation.
3. Computational solutions for preferred orientation bias, such as spIsoNet and cryoPROS, have enabled high-resolution reconstructions of datasets previously limited by severe orientation bias. These tools use deep learning to correct angular anisotropy and particle misalignment without requiring changes to specimen preparation.
4. Advanced denoising algorithms like Topaz-Denoise have improved signal-to-noise ratios by approximately 100-fold over raw images. This has led to faster acquisition times and higher throughput in cryoEM workflows, making high-resolution structural analysis more accessible.
5. In cryoET, tools such as IsoNet and TomoNet have addressed the missing-wedge problem and streamlined subtomogram averaging. IsoNet uses a U-Net architecture to correct missing-wedge artifacts and enhance SNRs, while TomoNet automates particle detection and averaging on flexible lattices.
6. Automated atomic model building using tools like ModelAngelo, DeepTracer, and CryoREAD has transformed density maps into interpretable biological structures. These AI-driven approaches have demonstrated high accuracy in protein and nucleic acid modeling, significantly reducing manual effort.
7. The future of AI in cryoEM holds great promise with the potential integration of large language models and multi-agent systems. These advancements could further automate and streamline workflows, making high-resolution structural biology more efficient and accessible.
📜Paper: arxiv.org/abs/2507.19565
#CryoEM #DeepLearning #StructuralBiology #AI #Proteomics #CryoET #Automation
1
4
923
29 Jul 2025
Review of Deep Learning Applications to Structural Proteomics Enabled by Cryogenic Electron Microscopy and Tomography
1. The integration of deep learning into cryoEM and cryoET workflows has revolutionized structural biology by addressing long-standing challenges such as low signal-to-noise ratios, preferred orientation artifacts, and missing-wedge problems. These advancements have led to remarkable achievements including near-atomic resolution reconstructions with minimal manual intervention.
2. Automated particle picking using convolutional neural networks (CNNs) like Topaz, crYOLO, and CryoSegNet has significantly improved efficiency and accuracy. CryoSegNet, in particular, combines a U-Net with a pre-trained foundation model to achieve state-of-the-art performance in particle detection and segmentation.
3. Computational solutions for preferred orientation bias, such as spIsoNet and cryoPROS, have enabled high-resolution reconstructions of datasets previously limited by severe orientation bias. These tools use deep learning to correct angular anisotropy and particle misalignment without requiring changes to specimen preparation.
4. Advanced denoising algorithms like Topaz-Denoise have improved signal-to-noise ratios by approximately 100-fold over raw images. This has led to faster acquisition times and higher throughput in cryoEM workflows, making high-resolution structural analysis more accessible.
5. In cryoET, tools such as IsoNet and TomoNet have addressed the missing-wedge problem and streamlined subtomogram averaging. IsoNet uses a U-Net architecture to correct missing-wedge artifacts and enhance SNRs, while TomoNet automates particle detection and averaging on flexible lattices.
6. Automated atomic model building using tools like ModelAngelo, DeepTracer, and CryoREAD has transformed density maps into interpretable biological structures. These AI-driven approaches have demonstrated high accuracy in protein and nucleic acid modeling, significantly reducing manual effort.
7. The future of AI in cryoEM holds great promise with the potential integration of large language models and multi-agent systems. These advancements could further automate and streamline workflows, making high-resolution structural biology more efficient and accessible.
📜Paper: arxiv.org/abs/2507.19565
#CryoEM #DeepLearning #StructuralBiology #AI #Proteomics #CryoET #Automation

1
2
597
4 Jul 2025
Multimodal deep learning integration of cryo-EM and AlphaFold3 for high-accuracy protein structure determination
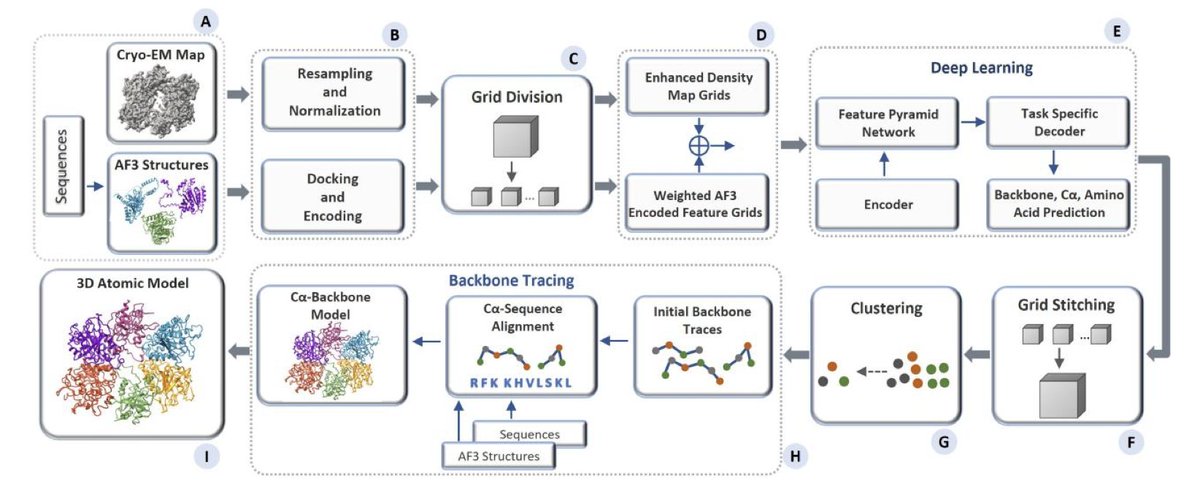
1.MICA introduces a fully automated deep learning method that integrates cryo-EM density maps and AlphaFold3 (AF3) predicted structures at both the input and output levels—enabling high-accuracy protein modeling without human intervention.
2.Unlike prior methods that only refine cryo-EM-based structures using AlphaFold predictions post hoc, MICA uses AF3 structures as input, fusing them with cryo-EM data from the start. This input-level integration is a key innovation that substantially boosts accuracy.
3.The architecture features a multimodal encoder-decoder framework with a Feature Pyramid Network (FPN), enabling hierarchical feature fusion and simultaneous prediction of backbone atoms, Cα atoms, and amino acid types.
4.MICA significantly outperforms state-of-the-art methods ModelAngelo and EModelX( AF) across multiple metrics (TM-score, Cα match, quality, sequence identity, and alignment length) on two benchmark datasets.
5.On the Cryo2StructData test set (average resolution 2.81 Å), MICA achieves a TM-score of 0.92, higher than EModelX( AF)’s 0.89 and ModelAngelo’s 0.75, and is the only method to reach high-accuracy modeling (TM > 0.9) on average.
6.It generates more complete and accurate models, with a 93.71% Cα match rate and an aligned Cα length 17.2% longer than ModelAngelo’s, while maintaining nearly identical or better sequence identity.
7.On the standard benchmark dataset (average resolution 3.17 Å), MICA again leads with a TM-score of 0.88 vs. EModelX( AF)'s 0.85 and ModelAngelo’s 0.57—54.4% higher than ModelAngelo’s.
8.MICA shows strong generalization: on 12 new cryo-EM density maps released after Jan 1, 2025 (test_2025), it achieved an average TM-score of 0.93 and >96% for all other metrics, demonstrating real-world robustness.
9.Its performance remains stable across a wide range of protein sizes and map resolutions. Regression analysis shows minimal accuracy degradation as resolution worsens, with MICA outperforming others in consistency.
10.The Cα match and TM-score are significantly higher for MICA due to its improved ability to predict a greater number of backbone atoms accurately, enabled by multimodal feature fusion and deep architectural optimization.
11.Limitations remain in chain/sequence registration in certain large complexes, particularly in low-resolution or noisy regions. Future directions include integrating better side-chain modeling and symmetry-aware sequence alignment.
12.Overall, MICA represents a step forward in automated cryo-EM-based protein modeling, capable of leveraging high-resolution experimental data and AF3 predictions in an end-to-end differentiable framework.
📜Paper: biorxiv.org/content/10.1101/…
#CryoEM #AlphaFold3 #ProteinStructure #DeepLearning #ComputationalBiology #Bioinformatics #StructuralBiology
2
6
836

6 Jun 2025
The headline is conformer diversity, but the sleeper win is full-length modeling of low-resolution transporters that stump other models:
CryoBoltz recovers four P-glycoprotein states with ~1.4 Å RMSD while ModelAngelo leaves up to 98% of residues blank (Figure 5 and Table 2)
1
4
474

3 Mar 2025
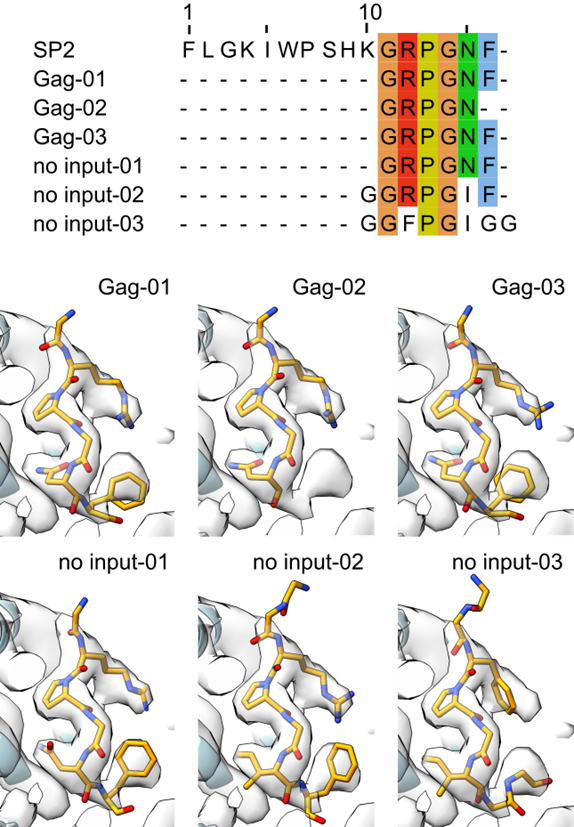
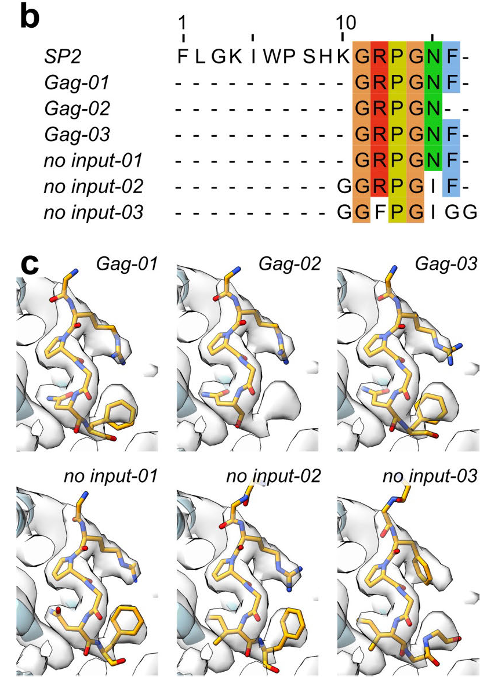
ModelAngelo from @SjorsScheres and @jamaliki1998
predicted that the side density is the C-term of the 16 AA long spacer peptide 2 (SP2) from HIV. Together with our experiments on Gag cleavage mutants, this was an indicator that the density is not a lipid but a peptide. 3/5.
1
3
26
3,896

9 Dec 2024
Visual Proteomics: 観測した構造から分子を決めに行く (ある意味 reverse genetics)。
ModelAngelo
nature.com/articles/s41586-0…
1
2
450
25 Nov 2024
Improved automated model building for cryo-EM maps using CryFold
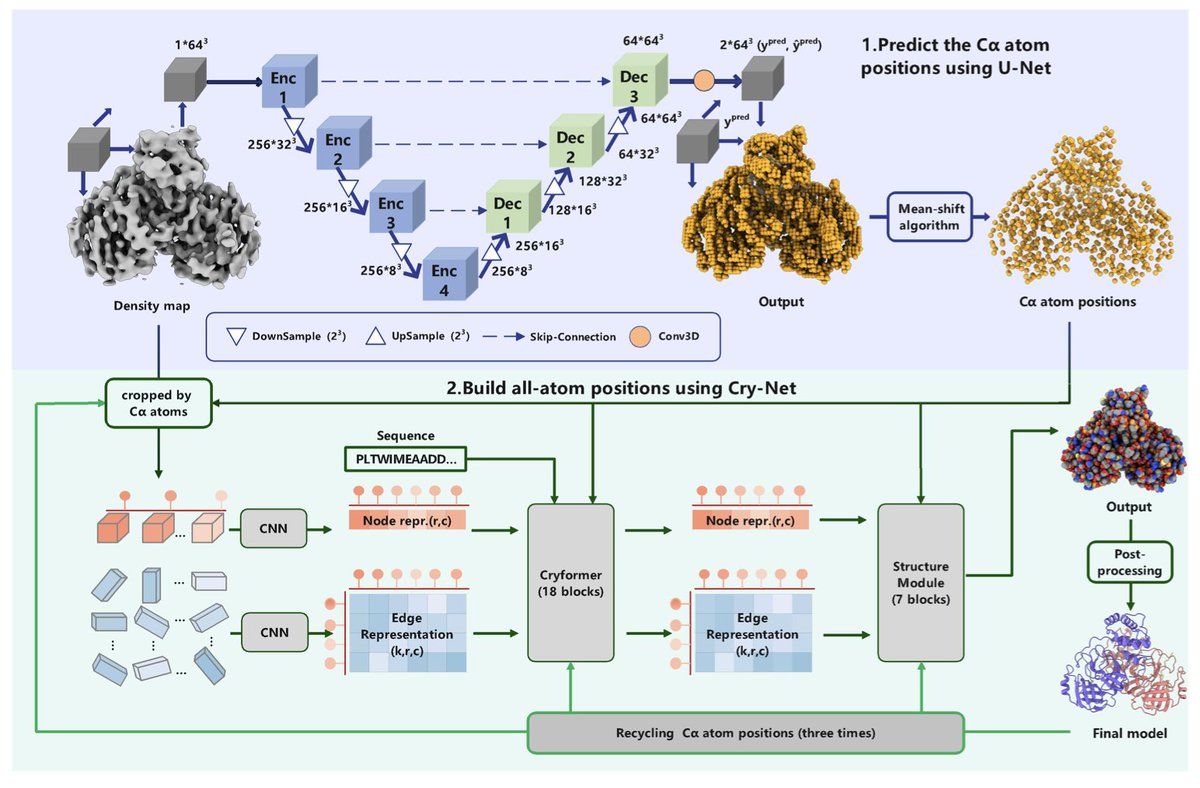
• CryFold, a novel tool for de novo model building from cryo-EM density maps, surpasses existing methods like ModelAngelo by integrating advancements from AlphaFold2 with unique adaptations such as 3D rotary position embedding and local attention mechanisms.
• Key achievements include modeling a 104-protein complex in just 5.6 hours, successfully detecting subtle conformational changes, and accurately differentiating paralogs in noisy map regions.
• Compared to ModelAngelo, CryFold provides higher completeness and accuracy in protein structure predictions, especially in low-resolution cryo-EM maps (below 4 Å resolution).
• Its unique 3D rotary position embedding ensures efficient spatial data encoding, enabling detailed structure refinement even in challenging regions of cryo-EM maps.
• CryFold can detect previously uncharacterized proteins directly from cryo-EM maps, as demonstrated in a photosynthetic complex where six new proteins were identified and modeled.
• This open-source tool also excels in segmenting density maps to isolate non-protein components, enhancing the understanding of molecular assemblies.
• CryFold’s efficient algorithms reduce GPU memory requirements, making high-resolution protein modeling accessible even on single GPUs.
• Applications range from resolving low-resolution regions to detecting novel proteins, showcasing its versatility in tackling modern structural biology challenges.
💻Code: github.com/SBQ-1999/CryFold
📜Paper: biorxiv.org/content/10.1101/…
#CryoEM #StructuralBiology #ProteinModeling #CryFold #AlphaFold2
16
52
4,735

13 Nov 2024
Thanks. You are right, they look very similar. We might have solved it. I'll keep you updated. Let's see what ModelAngelo thinks... If there is high resolution.
1
7
1,221
11 Nov 2024
Also, ModelAngelo from @SjorsScheres and @jamaliki1998 predicted that the ligand is the Ct part of the 16 AA long spacer peptide 2 from Gag. We could resolve the last 6 amino acids of SP2. The Ct of SP2 forms a bridge between two MA molecules triggering MA maturation. 6/x
1
4
348

29 Oct 2024
There we go🤷. We have #alphafold3, @alexjamesnoble's VirtualIce, a plethora of (nearly) automated 3D reconstruction tools, #ModelAngelo... and AIs that can generate hypotheses, test them and write papers. #cryoEM can go home I guess.
29 Oct 2024

In two new papers, researchers present AI agents that can do the entire job of a scientist – from hypothesis formulation, to literature research, data collection and analysis, all the way to writing a paper.
youtube.com/watch?v=BXSuaY9h…
1
1
5
1,270

18 Sep 2024
Excited to share our latest pre-print outlining a successful use case of #ModelAngelo and protein BLAST in helping us solve a tricky protein purification challenge in computational #ProteinDesign!
@UWproteindesign @UWBiochemistry @cryoEM_Papers
biorxiv.org/content/10.1101/…
3
6
13
1,030

28 Aug 2024
I was impressed by how well ModelAngelo performed on the unknown capsid. One can envision, a few years hence, a fully end-to-end software (wrapping concepts like SmartScope, Relion, and ModelAngelo) where you put a grid in and get a diagnostic result out.
1
1
7
371