
Multicopy Harderwijk neemt Kopieerderij over dlvr.it/TT3HGL
133

Jun 15
Quantum Nonlocality: Multicopy Resource Interconvertibility and Their Asymptotic Inequivalence journals.aps.org/prl/abstrac…
9

それです。培養条件を決めるのに1年以上かかった上に、遺伝子がクローニングできず(後でわかったのは当該遺伝子がmulticopy入ると致死的だった)、手を尽くしてクローンした。当時の技術で1.5kbの塩基配列を決めるのに6ヶ月かかり、決めてもイントロンが5つもあって読み枠がつながらず、、
1
3
32
14,062

New Research: Generation of iVero.219-mcRTA: a doxycycline-inducible high-titer KSHV producer cell line with multicopy orf50 integration frontiersin.org/articles/10.… #FrontiersIn #CellularInfectionMicrobiology
2
2
32

thrilled to share our joint work on the amplification of virulence in tan spot & ToxB @GourlieRyan @McdonaldMeganc @hafez_mnm answering to 25 yrs old Q on multicopy virulence gene | Molecular Plant-Microbe Interactions® apsjournals.apsnet.org/doi/a…
4
9
391

Apr 4
multicopy.pages.dev/
MultiCopy, a studying and productivity tool that allows users to copy and paste in bulk.
1
2
35

Evolutionary persistence of a highly prevalent multicopy mitochondrial-derived nuclear insertion (Mega-NUMT) in Neotropical ... biorxiv.org/content/10.64898… #biorxiv_evobio
2
373

Feb 25
Telomere consists of multicopy tandem repeat arrays and fully possesses all the essential features of a lifespan countdown timer / clock:
(1) It can be replicated during cell division to ensure each daughter cell inherits a copy;
(2) The shortened length of these arrays in somatic cells is replenished in germ cells, and germ cells must harbor the maximum copy number across all cells of an individual to sustain consumption throughout the entire life cycle, from embryonic development to maturation and senescence [31,22];
(3) The shortening rate of the arrays is malleable to adapt to the distinct developmental and senescent rates of different species and different cell types within the same species;
(4) In somatic cells, telomeres are consumed unidirectionally, enabling P53 to generate a concentration gradient along the time axis and form a temporal scale;
(5) All external factors or intrinsic genes that modulate the rate of senescence exert their effects by altering the shortening rate of these arrays;
(6) Given that some organisms can live for over a century—for instance, the Greenland shark has a lifespan of up to 400 years—the candidate driver substance that propels the operation of the genetic program (analogous to the sand in an hourglass) must be extremely stable and lack a half-life. In contrast, proteins, RNA, mitochondrial DNA (mtDNA), and chemical modifications of DNA and histones are highly unstable, have distinct half-lives, and exist in a dynamic equilibrium of constant degradation and replenishment. For example, DNA methylation and demethylation, as well as histone acetylation and deacetylation, occur simultaneously, making it impossible for these molecules to form a temporal measurement and precluding them from acting as a timer. In other words, the fundamental cause of aging does not lie in RNA, proteins, mtDNA, or epigenetic modifications, and research focused on these directions will never uncover the root cause of aging.
Thus, prior to the proposal of the TRCS model, among the numerous theories of cellular senescence, only the telomere theory is valid. In the causal relationship underlying cellular senescence, if telomere shortening is the cause of cellular senescence, then elongating telomere length in senescent cells will inevitably lead to a significant increase in cell replication capacity, accompanied by the reversion to a youthful cellular phenotype. Conversely, if telomere shortening is merely a consequence of cellular senescence, elongating telomere length in senescent cells will not enhance replication capacity nor restore a youthful state. In 2015, scientists at Stanford University first extended the telomere length of skin stem cells by 0.9 kb using hTERT mRNA, which increased the cell replication number by 28 passages [46]; Bodnar (1998) introduced the hTERT gene into human retinal pigment epithelial cells and foreskin fibroblasts, and observed a reduction in the senescence marker β-galactosidase, along with a marked increase in cell replication capacity [47].
Notably, telomere elongation leading to a significant boost in cell replication capacity is an effect that cannot be achieved by any other aging models or intervention strategies.
Further read: Huang, Bilu, The Information Theory of Aging Is Not Reliable (June 21, 2025). Available at SSRN: ssrn.com/abstract=5315671
Feb 19

“My research focus starts from a clear premise: aging as a genetically programmed phenomenon, executed by the epigenome.”
That the epigenome serves as the execution layer is largely non-controversial at this point. But what/where is the “clock” that initiates and regulates this?
1
2
15
657
Feb 25
Telomere consists of multicopy tandem repeat arrays and fully possesses all the essential features of a lifespan countdown timer / clock:
(1) It can be replicated during cell division to ensure each daughter cell inherits a copy;
(2) The shortened length of these arrays in somatic cells is replenished in germ cells, and germ cells must harbor the maximum copy number across all cells of an individual to sustain consumption throughout the entire life cycle, from embryonic development to maturation and senescence [31,22];
(3) The shortening rate of the arrays is malleable to adapt to the distinct developmental and senescent rates of different species and different cell types within the same species;
(4) In somatic cells, telomeres are consumed unidirectionally, enabling P53 to generate a concentration gradient along the time axis and form a temporal scale;
(5) All external factors or intrinsic genes that modulate the rate of senescence exert their effects by altering the shortening rate of these arrays;
(6) Given that some organisms can live for over a century—for instance, the Greenland shark has a lifespan of up to 400 years—the candidate driver substance that propels the operation of the genetic program (analogous to the sand in an hourglass) must be extremely stable and lack a half-life. In contrast, proteins, RNA, mitochondrial DNA (mtDNA), and chemical modifications of DNA and histones are highly unstable, have distinct half-lives, and exist in a dynamic equilibrium of constant degradation and replenishment. For example, DNA methylation and demethylation, as well as histone acetylation and deacetylation, occur simultaneously, making it impossible for these molecules to form a temporal measurement and precluding them from acting as a timer. In other words, the fundamental cause of aging does not lie in RNA, proteins, mtDNA, or epigenetic modifications, and research focused on these directions will never uncover the root cause of aging.
Thus, prior to the proposal of the TRCS model, among the numerous theories of cellular senescence, only the telomere theory is valid. In the causal relationship underlying cellular senescence, if telomere shortening is the cause of cellular senescence, then elongating telomere length in senescent cells will inevitably lead to a significant increase in cell replication capacity, accompanied by the reversion to a youthful cellular phenotype. Conversely, if telomere shortening is merely a consequence of cellular senescence, elongating telomere length in senescent cells will not enhance replication capacity nor restore a youthful state. In 2015, scientists at Stanford University first extended the telomere length of skin stem cells by 0.9 kb using hTERT mRNA, which increased the cell replication number by 28 passages [46]; Bodnar (1998) introduced the hTERT gene into human retinal pigment epithelial cells and foreskin fibroblasts, and observed a reduction in the senescence marker β-galactosidase, along with a marked increase in cell replication capacity [47].
Notably, telomere elongation leading to a significant boost in cell replication capacity is an effect that cannot be achieved by any other aging models or intervention strategies.
Further read: Huang, Bilu, The Information Theory of Aging Is Not Reliable (June 21, 2025). Available at SSRN: ssrn.com/abstract=5315671
2
37
Feb 20
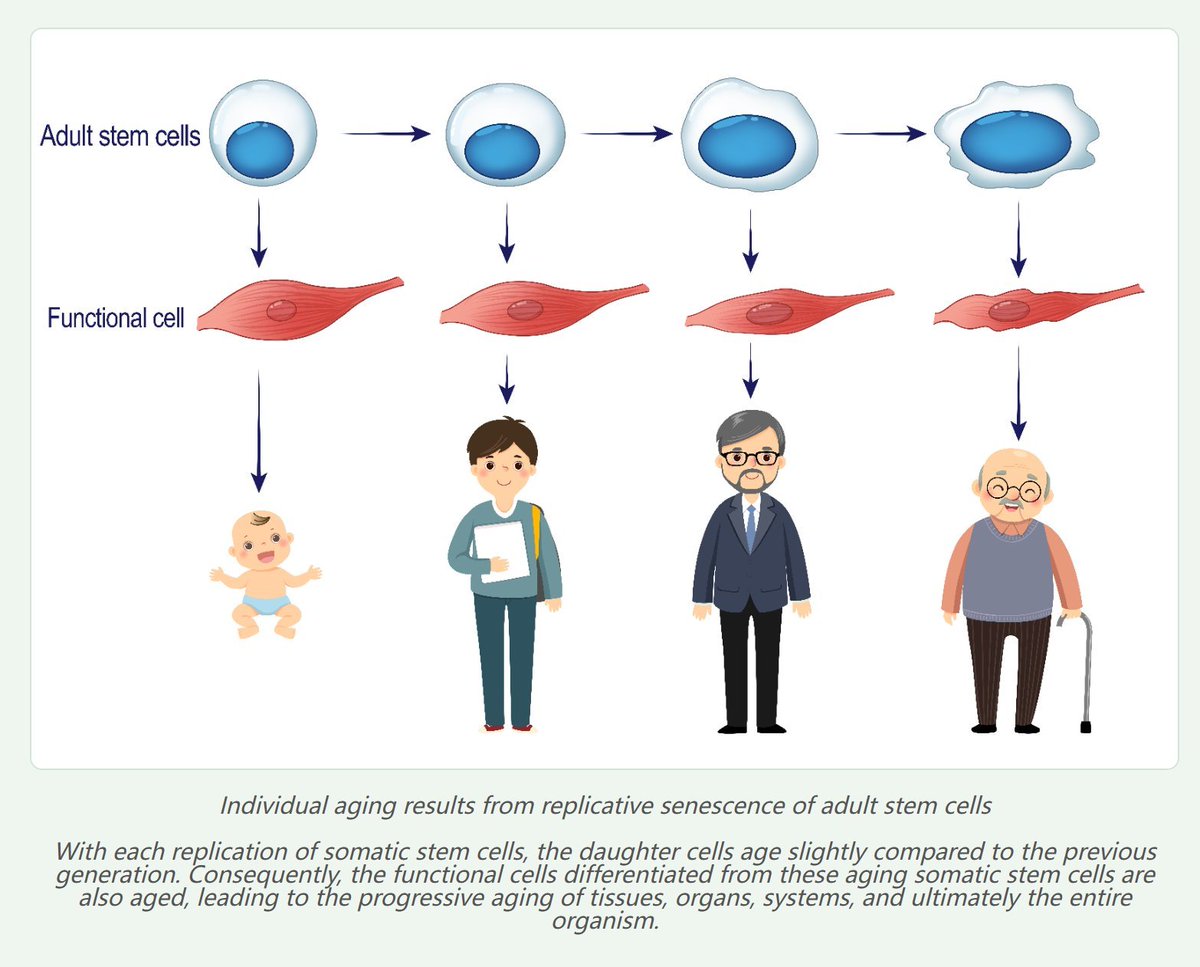
💯I published the stem cell theory of individual aging as early as 1998. The main content of this theory is as follows:Various tissues and organs in the human body are composed of two major types of cells—adult stem cells and functional cells.Every day, some functional cells and adult stem cells die due to various causes, such as DNA mutations, viral infections, and cellular physiological senescence.The lost functional cells are replenished by the differentiation of adult stem cells, while the lost adult stem cells are replenished by the self‑renewal of adult stem cells.
However, adult stem cells also undergo physiological senescence caused by the shortening of multicopy repetitive DNA such as telomeres.The functional cells differentiated and replenished from senescent adult stem cells are also senescent, leading to cascading aging of tissues, organs, and the whole organism.Therefore, the primary cause of individual aging is not the reduction in the number of adult stem cells, but the senescence of adult stem cells themselves.
Based on this, I propose that transplanting young autologous adult stem cells into aged individuals, or reversing the senescence of endogenous adult stem cells in the tissues of an individual, can greatly extend lifespan and even achieve rejuvenation.

Feb 20

And just a comment on "why do telomeres matter for post mitotic tissue", I believe the telomere length of stem cells are what is most important, not the telomere length of differentiated cells.
3
2
20
1,023
Feb 20
💯I published the stem cell theory of individual aging as early as 1998. The main content of this theory is as follows:Various tissues and organs in the human body are composed of two major types of cells—adult stem cells and functional cells.Every day, some functional cells and adult stem cells die due to various causes, such as DNA mutations, viral infections, and cellular physiological senescence.The lost functional cells are replenished by the differentiation of adult stem cells, while the lost adult stem cells are replenished by the self‑renewal of adult stem cells.
However, adult stem cells also undergo physiological senescence caused by the shortening of multicopy repetitive DNA such as telomeres.The functional cells differentiated and replenished from senescent adult stem cells are also senescent, leading to cascading aging of tissues, organs, and the whole organism.Therefore, the primary cause of individual aging is not the reduction in the number of adult stem cells, but the senescence of adult stem cells themselves.
Based on this, I propose that transplanting young autologous adult stem cells into aged individuals, or reversing the senescence of endogenous adult stem cells in the tissues of an individual, can greatly extend lifespan and even achieve rejuvenation.

2
3
42
Feb 20
Specifically, I proposed around 2011 that multicopy repetitive DNA such as telomeres act as temporal drivers for the programmed gene expression during cellular senescence. The chromosomal genome can be regarded as a database, while multicopy repetitive DNA including telomeres serves as the index of this database. The underlying regulatory mechanism may primarily involve the regulation of DNA methylation drift by modulating the level of the tumor suppressor protein P53. Details are presented as follows:
Theory of the Genetic Program Driver for Aging
The core content of the Theory of the Genetic Program Driver for Aging [4–5] is as follows: All organisms have a relatively fixed timetable of development, maturation, senescence, and death, which belongs to an ordered and programmed process. Therefore, the essence of aging is the staged temporal expression of three major gene groups: early-, mid-, and late-expressed genes. This is consistent with a study published in Nature Medicine, which used blood testing to reveal that human aging does not progress uniformly and gradually; instead, there likely exist physiological turning points where “quantitative changes lead to qualitative changes” [6].
The functions of the three gene groups are:
1. The early gene group is mainly responsible for embryonic development.
2. The mid gene group primarily supports survival and reproduction.
3. The late gene group mainly functions to disrupt normal physiology.
Taking terminally differentiated cells such as hepatocytes as an example:
During the embryonic period, hepatocytes mainly express early genes such as alpha-fetoprotein (AFP). In childhood and adulthood, AFP expression is silenced, and mid genes such as albumin are expressed instead. In old age, albumin expression gradually ceases, and late genes such as urinary protein [7] become activated.
Thus, the essence of aging is the sequential activation and silencing of specific gene groups.
Since hepatocytes can resume AFP synthesis during carcinogenesis, the age‑driven sequential gene expression is reversible. In other words, this mode of gene regulation differs from that of cell differentiation, which is generally stable and irreversible. For instance, malignant transformation of hepatocytes does not convert them into skin cells or other tissue‑type cells.
Many theories have proposed that aging is a genetic program, but none have clarified how such a program operates. Given that the genome remains largely unchanged throughout an individual’s lifespan, a critical question arises: how can fixed genes drive programmed gene expression?
I propose that a temporal driver is required to guide gene expression in a time‑dependent manner, much like a drive mechanism moves film in a movie projector or tape in a computer.
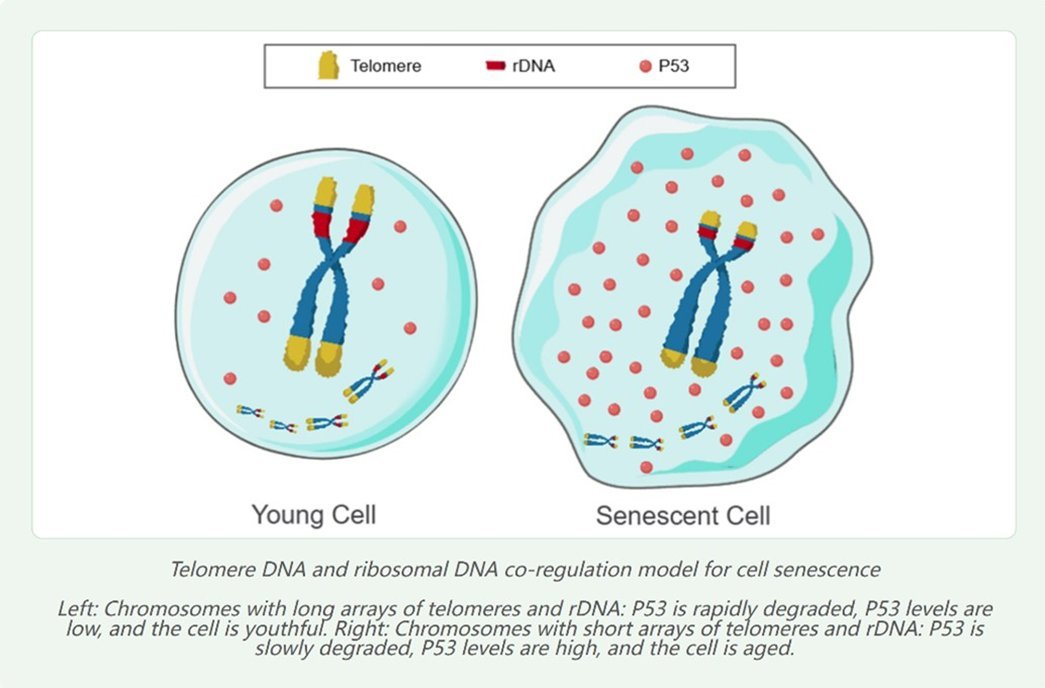
Because the copy number of multicopy DNA sequences such as telomeres decreases with cellular senescence, I hypothesize that multicopy repetitive DNA including telomeres act as the temporal driver of programmed gene expression during cellular senescence. The chromosomal genome can be regarded as a database, while multicopy repetitive DNA such as telomeres serve as the index of this database.
The specific regulatory mechanism likely involves the modulation of DNA methylation drift by regulating the level of the tumor suppressor protein p53.
Supporting evidence includes:
1. Facioscapulohumeral muscular dystrophy (FSHD) is a hereditary muscle disorder mainly caused by expression of the DUX4 gene, which is silenced in young individuals. In 2013, Guido Stadler from the University of Texas Southwestern Medical Center reported that shorter telomeres correlate with higher DUX4 expression, with activity increasing up to 10‑fold as telomeres shorten.
2. A study by the research group of Kathryn Demanelis at the University of Chicago, published in Science, revealed that telomere length is associated with the expression levels of more than 1,000 genes [8].
3. When activated, p53 influences the expression of thousands of genes [Andrysik Z, et al. Genome Research, 2017, 27(10): 1645–1657].
Because telomere shortening can elevate p53 levels, telomeres may regulate the expression of thousands of genes via p53 mediation.
References
[4] 黄必录.衰老的生命周期程序驱动学说[J].中国老年病杂志,2011,8:167-185.
Huang Bilu. The Life Cycle Driving Theory of Aging. Chinese Journal of Geriatrics, 2011, 8: 167–185.
[5] 黄必录,个体衰老与干细胞应用 颠覆传统理论,揭示衰老原因[C].中国老年学学会衰老与抗衰老科学委员会,第二届全国衰老与抗衰老学术大会论文集,浙江金华,金华市医学会,2009,112-115.
Huang Bilu. Individual aging and stem cell application: subverting traditional theories and revealing the causes of aging. In: Proceedings of the 2nd National Conference on Aging and Anti-Aging, Chinese Gerontological Society, Jinhua, Zhejiang, 2009: 112–115.
1
4
232
Feb 18
Metabolism can also promote telomere transcription. As the amount of RNA transcribed from telomeric DNA increases, the rate of telomere shortening accelerates when RNA levels near telomeres rise. Since telomeric DNA and rDNA are multicopy tandem repeat sequences with inherently poor stability, histones must be stripped during transcription or replication to expose the DNA and unwind the double-stranded helix. At this stage, they are highly vulnerable to various damages caused by reactive oxygen species and the transcription process, leading to the shortening of telomeres and rDNA arrays in cells.
2
50
Feb 1
According to the TRCS aging model, the fundamental mechanism determining the rate of aging is the rate of shortening of telomeres and/or ribosomal DNA (rDNA). Although mouse telomeres (50 kb) are several times longer than human telomeres (9 kb), their rate of shortening is approximately 100 times faster. Factors influencing the rate of aging, such as genes, species, body temperature, stress, exercise, and calorie restriction (CR), all exert their effects by modulating the rate of shortening of telomeres and/or rDNA.
For example, CR has been shown to reduce the rate of telomere shortening in rats (Pendergrass et al., 2001) and mice (Vera et al., 2013). Excessive food intake in Drosophila also increases the rate of rDNA shortening. This is because telomeric DNA and rDNA are multicopy tandem repeat sequences with inherently poor stability. During transcription or replication, histones must be stripped, exposing the DNA and unwinding the double helix, rendering it highly susceptible to damage and disruption from various factors, leading to copy loss. rDNA contraction leads to an increase in the concentration of the tumor suppressor protein p53, driving cells into a senescent state. Therefore, the fundamental reason why calorie restriction extends lifespan in animals is by slowing the rate of shortening of both telomeres and rDNA.
Mice have an initial telomere length of 50 kb, a telomere shortening rate of 6,420 bp per year, and a lifespan of 2.5–3 years. In humans, the initial telomere length is 15 kb, the annual shortening rate is 70 bp, and the lifespan is 75–85 years.
Goats have an annual telomere shortening rate of 363 bp and a lifespan of approximately 15 years; reindeer have an annual telomere shortening rate of 531 bp and a lifespan of 10–15 years; Audouin's gulls have an annual telomere shortening rate of 771 bp and a lifespan of 10–15 years; bottlenose dolphins have a telomere shortening rate of 766 base pairs per year and a lifespan of approximately 40 years; elephants have a cellular telomere shortening rate of 109 base pairs per year and a lifespan of approximately 60–70 years.
Furthermore, with advancing age, the regulatory role of rDNA in aging outweighs that of telomeres, which means that using telomeres alone to measure the rate of aging is inaccurate.
Jan 29

Lifespan is 50% heritable, twice previous estimates! In our new paper out today in @ScienceMagazine , we used mathematical modeling to remove extrinsic mortality, and validated with new twin data.
4
8
35
3,294
Jan 23
Telomeric DNA consists of multicopy tandem repeat arrays and fully possesses all the essential features of a lifespan countdown timer / clock:
(1) It can be replicated during cell division to ensure each daughter cell inherits a copy;
(2) The shortened length of these arrays in somatic cells is replenished in germ cells, and germ cells must harbor the maximum copy number across all cells of an individual to sustain consumption throughout the entire life cycle, from embryonic development to maturation and senescence [31,22];
(3) The shortening rate of the arrays is malleable to adapt to the distinct developmental and senescent rates of different species and different cell types within the same species;
(4) In somatic cells, telomeres are consumed unidirectionally, enabling P53 to generate a concentration gradient along the time axis and form a temporal scale;
(5) All external factors or intrinsic genes that modulate the rate of senescence exert their effects by altering the shortening rate of these arrays;
(6) Given that some organisms can live for over a century—for instance, the Greenland shark has a lifespan of up to 400 years—the candidate driver substance that propels the operation of the genetic program (analogous to the sand in an hourglass) must be extremely stable and lack a half-life. In contrast, proteins, RNA, mitochondrial DNA (mtDNA), and chemical modifications of DNA and histones are highly unstable, have distinct half-lives, and exist in a dynamic equilibrium of constant degradation and replenishment. For example, DNA methylation and demethylation, as well as histone acetylation and deacetylation, occur simultaneously, making it impossible for these molecules to form a temporal measurement and precluding them from acting as a timer. In other words, the fundamental cause of aging does not lie in RNA, proteins, mtDNA, or epigenetic modifications, and research focused on these directions will never uncover the root cause of aging.
Thus, prior to the proposal of the TRCS model, among the numerous theories of cellular senescence, only the telomere theory is valid. In the causal relationship underlying cellular senescence, if telomere shortening is the cause of cellular senescence, then elongating telomere length in senescent cells will inevitably lead to a significant increase in cell replication capacity, accompanied by the reversion to a youthful cellular phenotype. Conversely, if telomere shortening is merely a consequence of cellular senescence, elongating telomere length in senescent cells will not enhance replication capacity nor restore a youthful state. In 2015, scientists at Stanford University first extended the telomere length of skin stem cells by 0.9 kb using hTERT mRNA, which increased the cell replication number by 28 passages [46]; Bodnar (1998) introduced the hTERT gene into human retinal pigment epithelial cells and foreskin fibroblasts, and observed a reduction in the senescence marker β-galactosidase, along with a marked increase in cell replication capacity [47].
Notably, telomere elongation leading to a significant boost in cell replication capacity is an effect that cannot be achieved by any other aging models or intervention strategies.
Experimental studies have also shown that a single tail vein injection of AAV9-mTERT in mice over their lifetime resulted in reduced DNA damage, improved glucose tolerance, alleviated cognitive decline, delayed tumorigenesis, and a 24% extension in the average lifespan of the mice [Bernardes de Jesus B, Vera E, Schneeberger K, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012 Aug;4(8):691-704. doi: 10.1002/emmm.201200245.].
However, due to the excessively long telomeres in mice, the lifespan-extending effect of telomerase therapy in mice may be weaker than that in humans. Furthermore, telomere shortening decelerates with age and eventually rebounds: for example, human telomere length increases in individuals aged 80–90 years [48-49], and telomere elongation has also been observed in senescent naked mole-rats [50]. Our own research has also demonstrated that a reduction in 45S rDNA copy number leads to elongated telomeres in white blood cells. Therefore, rDNA exerts a greater driving effect on aging than telomeres in the elderly, meaning that the lifespan-extending efficacy of telomere elongation via telomerase alone will gradually diminish. This finding nonetheless confirms the validity of the telomere theory of aging.
Finally, research shows that leukocyte telomere length is longer in 80-year-old elders than in 20-year-old young adults. However, measuring leukocyte telomere length does not reflect the actual physiological state. Our experimental data indicate that knockout of 45S rDNA leads to telomere elongation in white blood cells, demonstrating that increased leukocyte telomere length is a consequence of aging. Thus, only the measurement of telomere length in stem cells can accurately reflect the true biological state of an organism.
2
4
13
704
Jan 17
First, telomeric DNA consists of multicopy tandem repeat arrays and fully possesses all the essential features of a lifespan countdown timer:
(1) It can be replicated during cell division to ensure each daughter cell inherits a copy;
(2) The shortened length of these arrays in somatic cells is replenished in germ cells, and germ cells must harbor the maximum copy number across all cells of an individual to sustain consumption throughout the entire life cycle, from embryonic development to maturation and senescence [31,22];
(3) The shortening rate of the arrays is malleable to adapt to the distinct developmental and senescent rates of different species and different cell types within the same species;
(4) In somatic cells, telomeres are consumed unidirectionally, enabling p53 to generate a concentration gradient along the time axis and form a temporal scale;
(5) All external factors or intrinsic genes that modulate the rate of senescence exert their effects by altering the shortening rate of these arrays;
(6) Given that some organisms can live for over a century—for instance, the Greenland shark has a lifespan of up to 400 years—the candidate driver substance that propels the operation of the genetic program (analogous to the sand in an hourglass) must be extremely stable and lack a half-life. In contrast, proteins, RNA, mitochondrial DNA (mtDNA), and chemical modifications of DNA and histones are highly unstable, have distinct half-lives, and exist in a dynamic equilibrium of constant degradation and replenishment. For example, DNA methylation and demethylation, as well as histone acetylation and deacetylation, occur simultaneously, making it impossible for these molecules to form a temporal measurement and precluding them from acting as a timer. In other words, the fundamental cause of aging does not lie in RNA, proteins, mtDNA, or epigenetic modifications, and research focused on these directions will never uncover the root cause of aging.
Thus, prior to the proposal of the TRCS model, among the numerous theories of cellular senescence, only the telomere theory is valid. In the causal relationship underlying cellular senescence, if telomere shortening is the cause of cellular senescence, then elongating telomere length in senescent cells will inevitably lead to a significant increase in cell replication capacity, accompanied by the reversion to a youthful cellular phenotype. Conversely, if telomere shortening is merely a consequence of cellular senescence, elongating telomere length in senescent cells will not enhance replication capacity nor restore a youthful state. In 2015, scientists at Stanford University first extended the telomere length of skin stem cells by 0.9 kb using hTERT mRNA, which increased the cell replication number by 28 passages [46]; Bodnar (1998) introduced the hTERT gene into human retinal pigment epithelial cells and foreskin fibroblasts, and observed a reduction in the senescence marker β-galactosidase, along with a marked increase in cell replication capacity [47].
Notably, telomere elongation leading to a significant boost in cell replication capacity is an effect that cannot be achieved by any other aging models or intervention strategies.
Experimental studies have also shown that a single tail vein injection of AAV9-mTERT in mice over their lifetime resulted in reduced DNA damage, improved glucose tolerance, alleviated cognitive decline, delayed tumorigenesis, and a 24% extension in the average lifespan of the mice [Bernardes de Jesus B, Vera E, Schneeberger K, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012 Aug;4(8):691-704. doi: 10.1002/emmm.201200245.].
However, due to the excessively long telomeres in mice, the lifespan-extending effect of telomerase therapy in mice may be weaker than that in humans. Furthermore, telomere shortening decelerates with age and eventually rebounds: for example, human telomere length increases in individuals aged 80–90 years [48-49], and telomere elongation has also been observed in senescent naked mole-rats [50]. Our own research has also demonstrated that a reduction in 45S rDNA copy number leads to elongated telomeres in white blood cells. Therefore, rDNA exerts a greater driving effect on aging than telomeres in the elderly, meaning that the lifespan-extending efficacy of telomere elongation via telomerase alone will gradually diminish. This finding nonetheless confirms the validity of the telomere theory of aging.
Finally, the figure provided shows that leukocyte telomere length is longer in 80-year-old elders than in 20-year-old young adults. However, measuring leukocyte telomere length does not reflect the actual physiological state. Our experimental data indicate that knockout of 45S rDNA leads to telomere elongation in white blood cells, demonstrating that increased leukocyte telomere length is a consequence of aging. Thus, only the measurement of telomere length in stem cells can accurately reflect the true biological state of an organism.
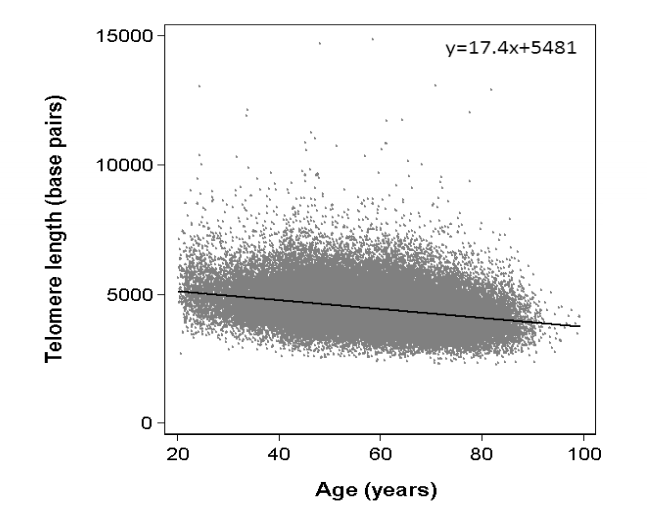
3
5
15
511
18 Dec 2025
All eukaryotes have telomeres and rDNA, both of which are multicopy tandem repeat sequences. Both telomeres and rDNA bind to the p53 protein. Therefore, telomeres and rDNA have universal applicability as countdown timers for cell replication and aging.
For example, the copy number of rDNA determines the replicative lifespan of yeast cells [Hotz M, Thayer NH, Hendrickson DG, et al. rDNA array length is a major determinant of replicative lifespan in budding yeast. Proc Natl Acad Sci U S A. 2022 Apr 12;119(15):e2119593119. doi: 10.1073/pnas.2119593119.], the rDNA in Drosophila shortens by half over its 40-day lifespan [Lu KL, Nelson JO, Watase GJ, et al. Transgenerational dynamics of rDNA copy number in Drosophila male germline stem cells. Elife. 2018 Feb 13;7:e32421. doi: 10.7554/eLife.32421.], and the shortening of telomeres and rDNA in mice and humans occurs at comparable levels [Ren R, Deng L, Xue Y, et al. Visualization of aging-associated chromatin alterations with an engineered TALE system. Cell Res. 2017 Apr;27(4):483-504. doi: 10.1038/cr.2017.18.].
Of course, the rate of aging is related to the shortening rate of telomeres and rDNA arrays and is influenced by numerous genetic and environmental factors, but is independent of the initial length of these arrays.
For example, mice have an initial telomere length of 50 kb, a telomere shortening rate of 6,420 bp per year, and a lifespan of 2.5-3 years; humans have 15 kb, an annual shortening rate of 70 bp, and a lifespan of 75-85 years; goats have an annual telomere shortening rate of 363 bp and a lifespan of about 15 years; reindeer have an annual shortening rate of 531 bp and a lifespan of 10-15 years; Audouin's gull has an annual shortening rate of 771 bp and a lifespan of 10-15 years; bottlenose dolphins have an annual telomere shortening rate of 766 base pairs and a lifespan of about 40 years; and elephants have an annual cellular telomere shortening rate of 109 base pairs and a lifespan of about 60 to 70 years.
It is worth noting that telomere shortening gradually slows down with age until it rebounds. For instance, telomeres lengthen in old naked mole-rats [Leonida S, Bennett N C, Leitch AR, et al. Patterns of telomere length with age in African mole-rats: New insights from quantitative fluorescence in situ hybridisation (qFISH)[J]. PeerJ, 2020, 8(13):e10498. doi: 10.7717/peerj.10498.]. The telomere length of a species of petrel increases with age: [Haussmann MF, Winkler DW, O'Reilly KM, Huntington CE, Nisbet ICT, Vleck CM. Telomeres shorten more slowly in long-lived birds and mammals than in short-lived ones. Proc R Soc Lond B Biol Sci[J], 2003, 270:1387-1392.] and [Mark F. Haussmann, Robert A. Mauck. Telomeres and Longevity: Testing an Evolutionary Hypothesis. Molecular Biology and Evolution[J], 2008, 25(1):220–228, doi.org/10.1093/molbev/msm24…]. In their 2015 paper "Automated Assay of Telomere Length Measurement and Informatics for 100,000 Subjects," Kyle Lapham, Elizabeth H. Blackburn, et al. pointed out that telomere length shortens in both men and women with age. From age 50 to 75, men experience a greater reduction in telomere length than women. However, from age 80 to 90, telomere length slightly increases in both men and women [Lapham K, Kvale MN, Lin J, et al. Automated Assay of Telomere Length Measurement and Informatics for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics. 2015 Aug;200(4):1061-72. doi: 10.1534/genetics.115.178624.].
The paradoxical lengthening of telomeres in somatic tissues of the very old: aging effect meets birth-cohort effect [Stindl R. The Paradoxical Lengthening of Telomeres in Somatic Tissues of the Very Old: Aging Effect Meets Birth-Cohort Effect. J Exp Zool B Mol Dev Evol. 2016 Jun;326(4):213-4. doi: 10.1002/jez.b.22677.].
Our research also unexpectedly found that reducing the copy number of 45S rDNA leads to an increase in telomere length in T cells and NK cells. This may be a compensatory mechanism, perhaps explaining why telomeres lengthen with age in the rhizome of ginseng, ginkgo trees, pine trees, naked mole-rats (with telomeres of 16 kb), Leach's storm petrels, and the very elderly. It may also explain why Arabidopsis thaliana with knocked-out telomerase exhibits an extended lifespan and why mice show reduced lifespan or defects only in the third or fourth generation after knockout of the telomerase gene.
Since the role of rDNA in regulating aging becomes more significant than that of telomeres with increasing age, using telomeres alone to measure the rate of aging is inaccurate.
17 Dec 2025

multiple species analysis does not support aging as caused by telomere shortening or deficiencies in ribosomal DNA arrays
1
2
13
6,755
18 Dec 2025
All eukaryotes have telomeres and rDNA, both of which are multicopy tandem repeat sequences. Both telomeres and rDNA bind to the p53 protein. Therefore, telomeres and rDNA have universal applicability as countdown timers for cell replication and aging.
For example, the copy number of rDNA determines the replicative lifespan of yeast cells [Hotz M, Thayer NH, Hendrickson DG, et al. rDNA array length is a major determinant of replicative lifespan in budding yeast. Proc Natl Acad Sci U S A. 2022 Apr 12;119(15):e2119593119. doi: 10.1073/pnas.2119593119.], the rDNA in Drosophila shortens by half over its 40-day lifespan [Lu KL, Nelson JO, Watase GJ, et al. Transgenerational dynamics of rDNA copy number in Drosophila male germline stem cells. Elife. 2018 Feb 13;7:e32421. doi: 10.7554/eLife.32421.], and the shortening of telomeres and rDNA in mice and humans occurs at comparable levels [Ren R, Deng L, Xue Y, et al. Visualization of aging-associated chromatin alterations with an engineered TALE system. Cell Res. 2017 Apr;27(4):483-504. doi: 10.1038/cr.2017.18.].
Of course, the rate of aging is related to the shortening rate of telomeres and rDNA arrays and is influenced by numerous genetic and environmental factors, but is independent of the initial length of these arrays.
For example, mice have an initial telomere length of 50 kb, a telomere shortening rate of 6,420 bp per year, and a lifespan of 2.5-3 years; humans have 15 kb, an annual shortening rate of 70 bp, and a lifespan of 75-85 years; goats have an annual telomere shortening rate of 363 bp and a lifespan of about 15 years; reindeer have an annual shortening rate of 531 bp and a lifespan of 10-15 years; Audouin's gull has an annual shortening rate of 771 bp and a lifespan of 10-15 years; bottlenose dolphins have an annual telomere shortening rate of 766 base pairs and a lifespan of about 40 years; and elephants have an annual cellular telomere shortening rate of 109 base pairs and a lifespan of about 60 to 70 years.
It is worth noting that telomere shortening gradually slows down with age until it rebounds. For instance, telomeres lengthen in old naked mole-rats [Leonida S, Bennett N C, Leitch AR, et al. Patterns of telomere length with age in African mole-rats: New insights from quantitative fluorescence in situ hybridisation (qFISH)[J]. PeerJ, 2020, 8(13):e10498. doi: 10.7717/peerj.10498.]. The telomere length of a species of petrel increases with age: [Haussmann MF, Winkler DW, O'Reilly KM, Huntington CE, Nisbet ICT, Vleck CM. Telomeres shorten more slowly in long-lived birds and mammals than in short-lived ones. Proc R Soc Lond B Biol Sci[J], 2003, 270:1387-1392.] and [Mark F. Haussmann, Robert A. Mauck. Telomeres and Longevity: Testing an Evolutionary Hypothesis. Molecular Biology and Evolution[J], 2008, 25(1):220–228, doi.org/10.1093/molbev/msm24…]. In their 2015 paper "Automated Assay of Telomere Length Measurement and Informatics for 100,000 Subjects," Kyle Lapham, Elizabeth H. Blackburn, et al. pointed out that telomere length shortens in both men and women with age. From age 50 to 75, men experience a greater reduction in telomere length than women. However, from age 80 to 90, telomere length slightly increases in both men and women [Lapham K, Kvale MN, Lin J, et al. Automated Assay of Telomere Length Measurement and Informatics for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics. 2015 Aug;200(4):1061-72. doi: 10.1534/genetics.115.178624.].
The paradoxical lengthening of telomeres in somatic tissues of the very old: aging effect meets birth-cohort effect [Stindl R. The Paradoxical Lengthening of Telomeres in Somatic Tissues of the Very Old: Aging Effect Meets Birth-Cohort Effect. J Exp Zool B Mol Dev Evol. 2016 Jun;326(4):213-4. doi: 10.1002/jez.b.22677.].
Our research also unexpectedly found that reducing the copy number of 45S rDNA leads to an increase in telomere length in T cells and NK cells. This may be a compensatory mechanism, perhaps explaining why telomeres lengthen with age in the rhizome of ginseng, ginkgo trees, pine trees, naked mole-rats (with telomeres of 16 kb), Leach's storm petrels, and the very elderly. It may also explain why Arabidopsis thaliana with knocked-out telomerase exhibits an extended lifespan and why mice show reduced lifespan or defects only in the third or fourth generation after knockout of the telomerase gene.
Since the role of rDNA in regulating aging becomes more significant than that of telomeres with increasing age, using telomeres alone to measure the rate of aging is inaccurate.
5
132