
Jun 12
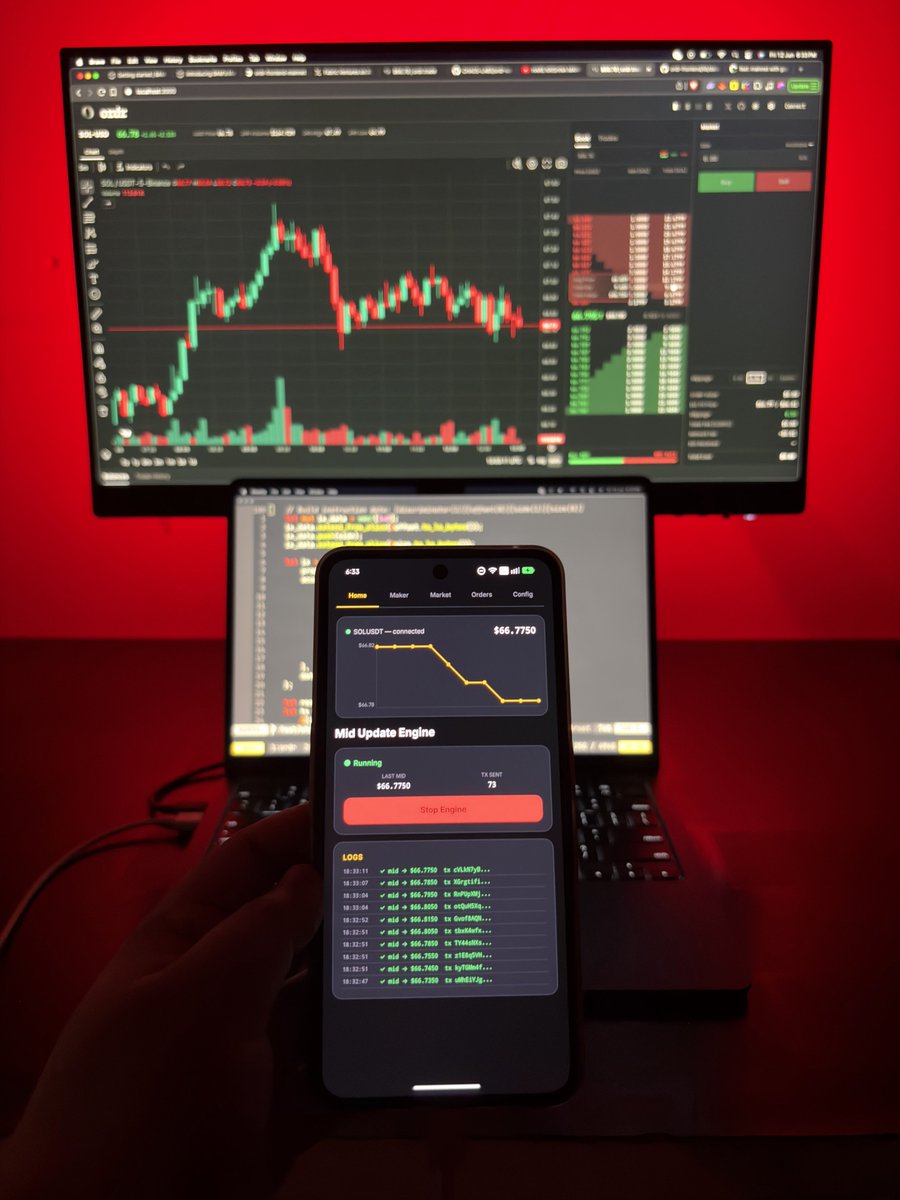
protocols keep market making behind closed doors. @ordrtrade opens it up. permissionless makers, onchain books, priority via jito BAM.
this is openMM. OPOS.
1
24
1,653

Jun 10
🧬 Master Molecular Dynamics! Join our 3-Day Hands-On Workshop on Python-Powered Molecular Simulations.
🔹 Learn OpenMM, MDTraj, & MDAnalysis
📅 July 27–31, 2026 ⏰ 7 PM IST | 8:30 AM CDT 💻 Live Hands-on
🔗 Register: forms.gle/9tMuFo5TwJS3tftQ6
#Bioinformatics #DrugDiscovery




15
hou zard retweeted

13 Aug 2024
Which protein binder design AI models will stand the test of time?
Here, we see:
EvoDiff ColabFold OpenMM HADDOCK for antibody discovery
biorxiv.org/content/10.1101/…
18
53
3,568



May 28
@ordrtrade made me research more on "propAMM vs openMM" !
- they finally opened the closed box - welcome to the openMM paradigm.
- flat 32 CU oracle update is crazy.


3
2
5
343

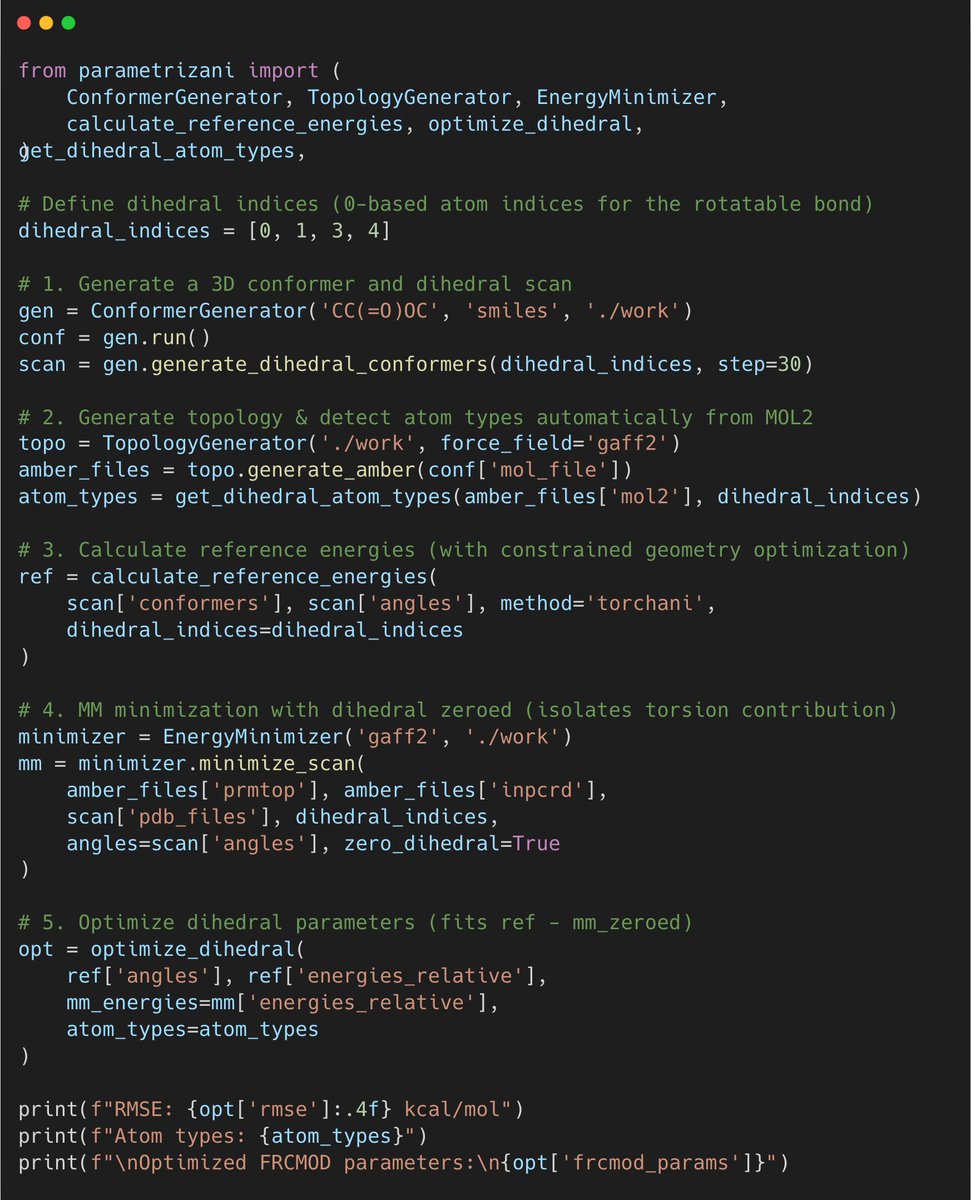
🚀 ParametrizANI is now a Python package!
What started as Google Colab notebooks is now a full pip-installable library for fast, accessible dihedral parametrization of small molecules, SMILES to AMBER/GROMACS/OpenMM in one script.
🧵 [1/3]
1
1
90

Scientific Agent Skills 是一个面向科研场景的 Agent 技能库。
它提供 138 个可直接使用的科研技能,可以让 Claude Code、Cursor、Codex 等 Agent 更好地处理科学研究、数据分析、文献写作和工程计算任务。
GitHub:github.com/K-Dense-AI/scient…
主要内容:
-138 个科研 Agent Skills
-覆盖生物信息学、药物发现、医学影像、机器学习、材料科学等方向
-支持 100 科研/金融数据库查询
-包含 RDKit、Scanpy、BioPython、OpenMM、Qiskit 等常用工具技能
-支持文献综述、科研写作、图表、海报、引用管理等任务
-可用于 Claude Code、Cursor、Codex、Gemini CLI 等 Agent
这个项目适合科研人员、数据分析师、AI 科研工具爱好者。

7
226
May 13
.@ordrtrade - open Market Making; openMM
A fully on chain order book exchange on Solana that gives market makers their own private accounts, cheap repricing, and protection from toxic arbitrage. The result: tighter spreads and better prices for everyone.

2
13
965
May 13
OpenMM - Open Market Making
May 13
OpenMM - Open Market Making
11
834

May 13
OpenMM - Open Market Making
May 13
OpenMM - Open Market Making
5
164

OpenMM - Open Market Making
May 13
OpenMM - Open Market Making
3
215

May 12
openMM - open market making - @ordrtrade
May 12

openMM - open market making - @ordrtrade
1
2
161

May 11
openMM era 🔥

2
15
623

May 6
一些例子:
k2.6:会只用 10 个 pose 做 docking / 用 mm gbsa 的结果告诉我某些变异导致了三个数量级的亲和度差异 / 经常(三分之二?)会编码出错后续自己检查出来然后返工 / 告诉我 openmm vs gromacs 关键指标虽然差了 10A 但是是“一致”的 等等 (2/n)
1
2
1,489

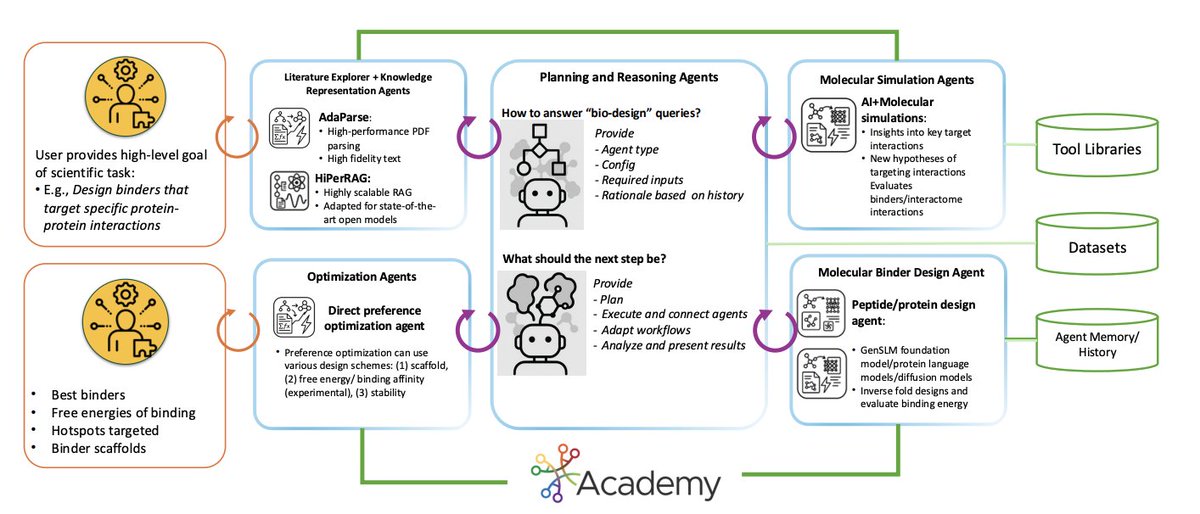
Scalable Agentic Reasoning for Designing Biologics Targeting Intrinsically Disordered Proteins
1. StructBioReasoner is a scalable multi-agent system aimed at designing biologics against intrinsically disordered proteins (IDPs), treating “what to design next” as an autonomous, HPC-orchestrated reasoning problem rather than a single-model generation task.
2. The key algorithmic idea is a tournament-based reasoning loop: specialized agents propose competing hypotheses (interfaces/hotspots, scaffolds, design methods), evaluate them with structure prediction simulation energetics, then promote winners and cull/annotate failures for the next round.
3. The system is explicitly built for IDP complexity: instead of assuming one stable target structure, it reasons over conformational heterogeneity and uses interactome-scale simulation to identify which interfaces are most plausible and therapeutically relevant to disrupt.
4. A major practical contribution is end-to-end tool orchestration on supercomputers via a federated agentic middleware (Academy), integrating workflow execution (Parsl) and remote/federated execution (Globus Compute) to scale design campaigns across heterogeneous HPC resources.
5. Literature grounding is treated as first-class infrastructure: HiPerRAG builds target-specific corpora (e.g., 1520 full-text papers for NMNAT-2), parses PDFs at scale (AdaParse), indexes embeddings (FAISS), and converts retrieved evidence into structured assertions a shared knowledge graph to reduce hallucinations.
6. Toolchain integration spans: structure prediction (Chai-1, Boltz-2x, plus PDB lookup), molecular dynamics (OpenMM; explicit/implicit solvent decisions), analysis (RMSD/RMSF/SASA/RoG; interaction energies), and binding estimation (MM-PBSA as a throughput/accuracy compromise for large campaigns).
7. Der f 21 benchmark (structured target): from 842 designed binders, 787 passed QC/verification; after MD MM-PBSA, 50.98% beat a literature human-designed reference binder (BindCraft binder 10) by a defined free-energy threshold, while most sequences remained novel (<30% identity vs prior BindCraft designs).
8. Der f 21 mechanistic consistency: simulations highlighted frequent contacts near known IgE-relevant epitope residues; designs often formed salt-bridge interactions (e.g., targeting E7), aligning computationally inferred interfaces with experimentally implicated allergen epitopes.
9. NMNAT-2 benchmark (IDR-rich, interactome-driven): the system simulated 18 interactome partners identified via RAG, prioritized interfaces dominated by IDR interactions, and recovered the well-studied NMNAT2:p53 interface as one binding mode among large-scale designed candidates.
10. NMNAT-2 scale and modes: 97,066 binders passed QC/structural validation (out of 266,606 generated), each run through explicit-solvent MD; analyses revealed three major binding modes, two targeting the IDR, and 84.5% of successful binders contacted at least one IDR residue—mirroring native interactome contact patterns.
11. Optimization is incorporated as an agent: preference pairs are constructed from multi-metric scoring (free energy, stability via RMSD/RMSF, developability features), enabling direct preference optimization (DPO) to bias future generation toward better candidates without full retraining.
12. Scaling results on Aurora (exascale-class system): MD agent showed robust weak scaling to 256 nodes (~26.6 µs/hour aggregate, ~80% efficiency vs 64-node baseline); binder design scaled to 512 nodes (efficiency drops beyond 256 nodes due to filesystem I/O); MM-PBSA became I/O-bound beyond 64 nodes, motivating staged filtering before expensive energetics.
💻Code: github.com/IDeA-ANL-ORNL/Str…
📜Paper: arxiv.org/abs/2512.15930
#ComputationalBiology #ProteinDesign #IDP #IntrinsicallyDisorderedProteins #AgenticAI #MultiAgentSystems #RAG #MolecularDynamics #HPC #Exascale #Biologics #DrugDiscovery
2
9
911