
Jun 10
1. 🚨 New Review: Protein Neddylation Beyond Tumor Cells — A Key Modulator of Anticancer Immunity 🔬🛡️
Neddylation isn't just about tumor cells; it actively shapes the tumor immune microenvironment! Targeting this pathway may overcome MLN4924 resistance.
Key Insights:
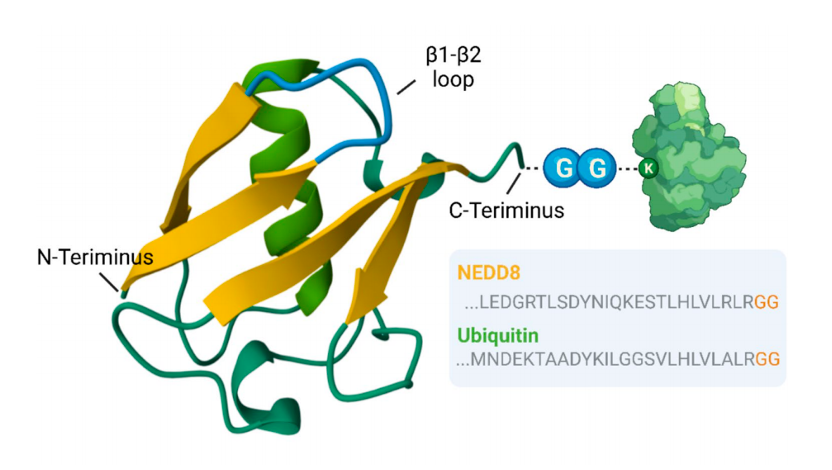
✅ Neddylation = ubiquitin-like modification via NAE → E2 → E3 cascade
✅ Primary substrates: Cullin-RING E3 ligases (regulate cell cycle, apoptosis, metabolism)
✅ MLN4924 = first-in-class NAE inhibitor, but faces drug resistance
✅ Neddylation modulates key immune cells (Table 1):
➡️ T cell differentiation/exhaustion, DC maturation, macrophage polarization
✅ Proposes a "Neddylation-Metabolite-Antitumor Immunity" regulatory axis
✅ Combining with immune checkpoint blockade may enhance MLN4924 efficacy
✅ Calls for targeted delivery systems to minimize off-target effects on immune cells
📚Authors: Xu et al.
📅Journal: *Phenomics*, 2026
📄Full study: doi.org/10.1007/s43657-025-0…
#Neddylation #ProteinModification #TumorImmunity #MLN4924 #Immunotherapy #DrugResistance #PrecisionMedicine #TumorMicroenvironment 🧬
16

28 Oct 2025
Learning the PTM Code through a Coarse-to-Fine, Mechanism-Aware Framework
1. A novel study introduces COMPASS-PTM, a novel framework that deciphers the complex post-translational modification (PTM) code by integrating evolutionary sequence information, physicochemical properties, and crosstalk dependencies in a coarse-to-fine learning approach. This method significantly advances the prediction of PTM sites and their corresponding regulatory enzymes, offering new insights into cellular signaling and disease mechanisms.
2. The COMPASS-PTM framework employs a two-stage architecture. The first stage, Multi-label Site Profiling Network (MSPN), performs proteome-scale PTM site detection by fusing protein language model features with chemical language model features. A key innovation is the crosstalk-aware prompting mechanism, which injects biological priors into the learning process to model inter-PTM dependencies, leading to more accurate and biologically coherent predictions.
3. The second stage, Enzyme-Substrate Pairing System (ESPS), links high-confidence PTM sites from the first stage to their regulatory enzymes. This stage integrates local substrate sequence motifs with global enzyme embedding representations to capture recognition specificity. The dual-gated residual fusion module effectively combines substrate and enzyme features, enhancing the model’s ability to predict enzyme-substrate interactions with high precision.
4. Across multiple proteome-scale benchmarks, COMPASS-PTM achieves state-of-the-art performance. The MSPN stage demonstrates a remarkable 122% relative F1-score improvement in multi-label site prediction on the dbPTM-ML dataset. The ESPS stage shows a 54% gain in zero-shot enzyme assignment on the DARKIN benchmark, confirming the effectiveness of the coarse-to-fine paradigm for generating mechanistically actionable insights.
5. Beyond quantitative performance, COMPASS-PTM offers qualitative advantages. It recovers canonical kinase motifs and predicts disease-associated PTM rewiring caused by missense variants. This interpretability grounded in biochemical mechanisms positions COMPASS-PTM as a robust and versatile resource for advancing PTM mechanistic studies and enabling novel biological discoveries.
6. The study also includes detailed ablation experiments that validate the contributions of each component of the COMPASS-PTM framework. The crosstalk-aware prompting module, the dual-modal fusion architecture, the hybrid loss function, and the dual-gated residual fusion module are all shown to be essential for the model’s superior performance, providing a clear rationale for the methodological choices.
7. The authors highlight the potential of COMPASS-PTM to extend beyond PTMs to decode other complex biological systems, such as enzymatic cascades and protein-protein interactions. By tightly coupling statistical learning with biochemical mechanisms, this mechanism-aware paradigm offers a generalizable route toward interpretable machine intelligence in computational biology.
📜Paper: arxiv.org/abs/2510.23492
#PTM #ProteinModification #MachineLearning #ComputationalBiology #Bioinformatics

1
4
15
1,376

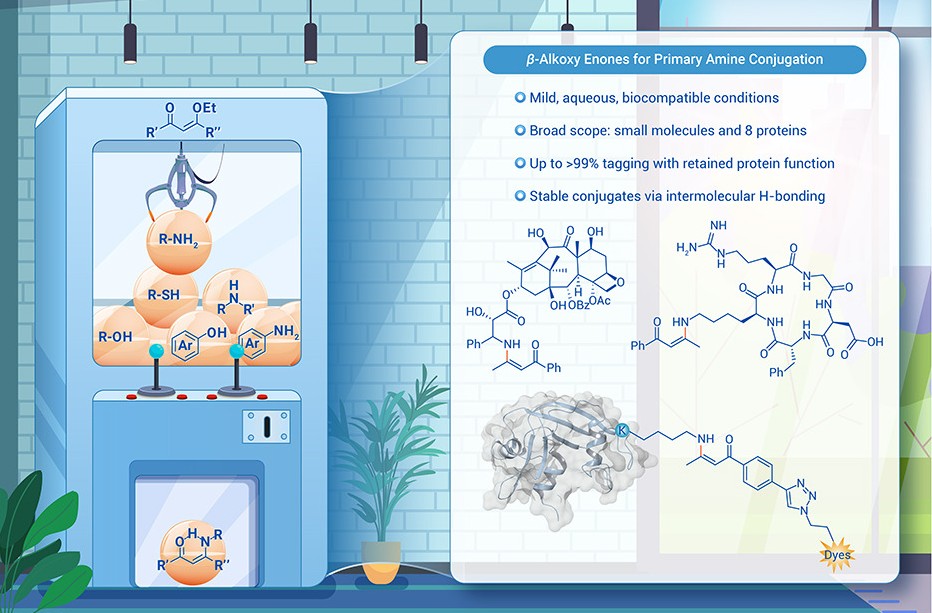
New in @The_InnovationJ! β-alkoxy enones for biocompatible primary amine conjugation.
This study on β-ethoxy enone-based amine conjugation heralds a significant advancement in chemical biology and pharmaceutical sciences, providing a novel, efficient approach for modifying biologically relevant molecules.
doi.org/10.1016/j.xinn.2025.…
#biocompatible #bioconjugation #ProteinModification #chemicalbiology
2
8
10,731
7 May 2025
MAP Format for Representing Chemical Modifications, Annotations, and Mutations in Protein Sequences: An Extension of the FASTA Format
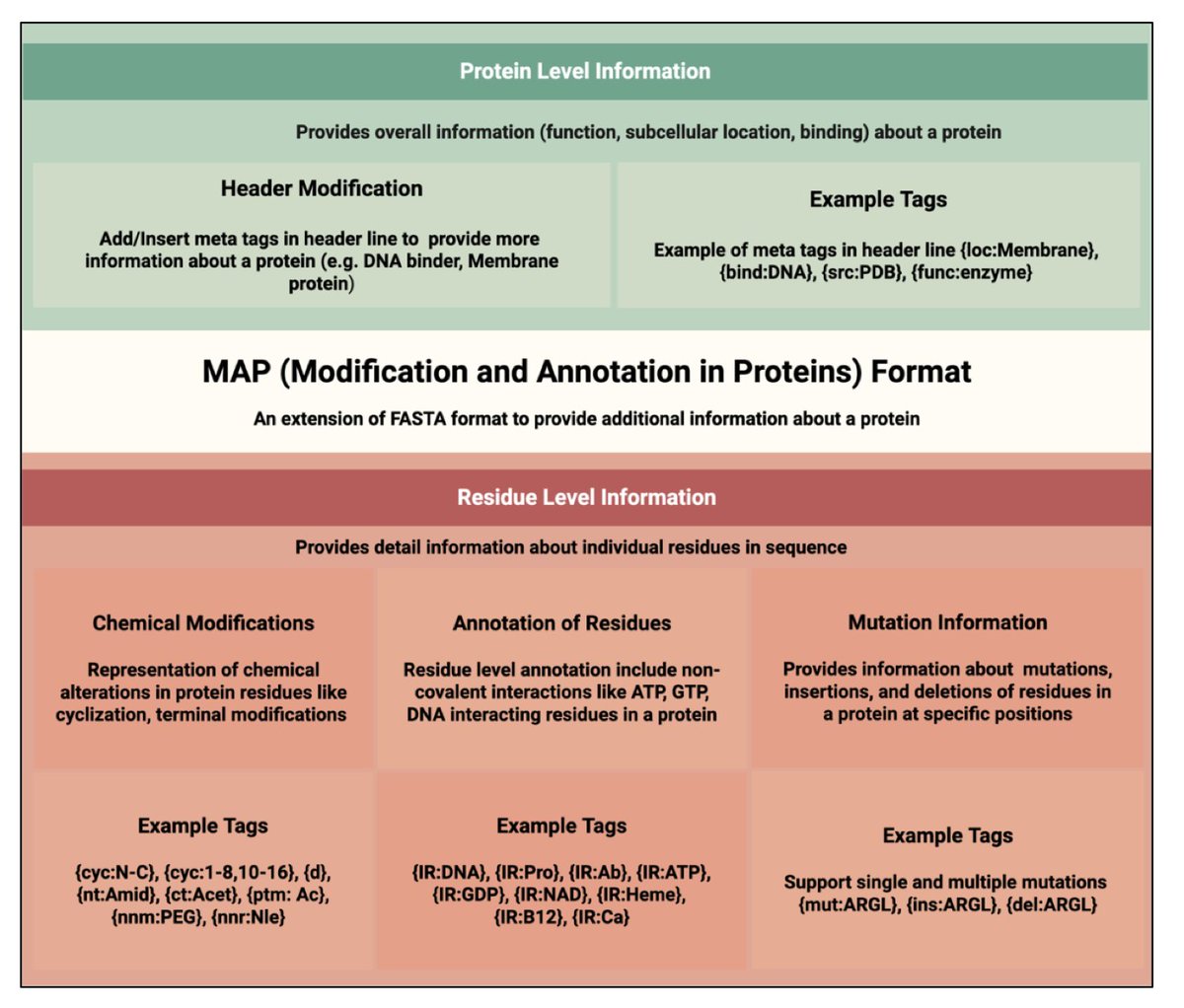
1. This study proposes MAP, a new sequence format that extends the widely used FASTA format to include in-line annotations for protein modifications, mutations, and structural features—retaining readability and compatibility with existing bioinformatics tools.
2. MAP uses intuitive curly-brace tags embedded directly within the protein sequence to denote modifications like phosphorylation, cyclization, non-natural residues, or D-amino acids, providing residue-level annotation in a single-line, human-readable format.
3. Header metadata is structured using tags like {org\:Homo sapiens} or {func\:DNA-binding}, allowing easy parsing of protein-level attributes like organism, subcellular location, and function—enhancing integration with protein databases.
4. Unlike PEFF, HELM, or BILN, which are either too simplistic or overly complex, MAP strikes a balance—simple enough for biologists to edit manually, yet expressive enough to represent therapeutic peptides and engineered proteins with chemical modifications.
5. MAP can denote sequence variants such as mutations (e.g., {mut\:R}), insertions ({ins\:MK}), and deletions ({del\:RTH}) within the linear sequence, enabling unified representation of wild-type and mutant forms for variant annotation and analysis.
6. The format supports tagging of interacting residues using {IR\:Target} (e.g., H{IR\:Zn}), enabling capture of functional sites like metal ion binding or nucleic acid interaction sites directly within the sequence.
7. Applications span protein therapeutics, synthetic biology, structural bioinformatics, and database curation. MAP is already positioned to enhance resources like UniProt, PDB, and peptide therapeutic databases by embedding rich residue-level context.
8. MAP is backward-compatible: removing all curly-brace tags produces a valid FASTA sequence, ensuring that legacy tools can still interpret core sequence content while MAP-aware tools leverage annotations.
9. The authors provide a web portal (MAPrepo) offering manuals, curated datasets, database exports, and Python scripts for converting between formats, facilitating immediate adoption by the community.
10. MAP’s simplicity, flexibility, and compatibility position it as a practical standard for next-generation protein sequence annotation—bridging raw sequences and chemically informed representations in a unified, FAIR-compliant format.
💻Code: webs.iiitd.edu.in/raghava/ma…
📜Paper: arxiv.org/abs/2505.03403
#ProteinSequence #FASTA #MAPformat #Bioinformatics #ProteinModification #PostTranslationalModifications #DataStandards #Proteomics #ComputationalBiology #SequenceAnnotation #TherapeuticProteins #FAIRdata

1
2
670
7 May 2025
MAP Format for Representing Chemical Modifications, Annotations, and Mutations in Protein Sequences: An Extension of the FASTA Format
1. This study proposes MAP, a new sequence format that extends the widely used FASTA format to include in-line annotations for protein modifications, mutations, and structural features—retaining readability and compatibility with existing bioinformatics tools.
2. MAP uses intuitive curly-brace tags embedded directly within the protein sequence to denote modifications like phosphorylation, cyclization, non-natural residues, or D-amino acids, providing residue-level annotation in a single-line, human-readable format.
3. Header metadata is structured using tags like {org\:Homo sapiens} or {func\:DNA-binding}, allowing easy parsing of protein-level attributes like organism, subcellular location, and function—enhancing integration with protein databases.
4. Unlike PEFF, HELM, or BILN, which are either too simplistic or overly complex, MAP strikes a balance—simple enough for biologists to edit manually, yet expressive enough to represent therapeutic peptides and engineered proteins with chemical modifications.
5. MAP can denote sequence variants such as mutations (e.g., {mut\:R}), insertions ({ins\:MK}), and deletions ({del\:RTH}) within the linear sequence, enabling unified representation of wild-type and mutant forms for variant annotation and analysis.
6. The format supports tagging of interacting residues using {IR\:Target} (e.g., H{IR\:Zn}), enabling capture of functional sites like metal ion binding or nucleic acid interaction sites directly within the sequence.
7. Applications span protein therapeutics, synthetic biology, structural bioinformatics, and database curation. MAP is already positioned to enhance resources like UniProt, PDB, and peptide therapeutic databases by embedding rich residue-level context.
8. MAP is backward-compatible: removing all curly-brace tags produces a valid FASTA sequence, ensuring that legacy tools can still interpret core sequence content while MAP-aware tools leverage annotations.
9. The authors provide a web portal (MAPrepo) offering manuals, curated datasets, database exports, and Python scripts for converting between formats, facilitating immediate adoption by the community.
10. MAP’s simplicity, flexibility, and compatibility position it as a practical standard for next-generation protein sequence annotation—bridging raw sequences and chemically informed representations in a unified, FAIR-compliant format.
💻Code: webs.iiitd.edu.in/raghava/ma…
📜Paper: arxiv.org/abs/2505.03403
#ProteinSequence #FASTA #MAPformat #Bioinformatics #ProteinModification #PostTranslationalModifications #DataStandards #Proteomics #ComputationalBiology #SequenceAnnotation #TherapeuticProteins #FAIRdata
2
549

18 Dec 2024
In a study led by @SIPBS_Strath, @OxfordStrubi and S-acylation experts from UK universities have received £6.4m to research the impact of #ProteinModification on cell physiology, which will advance the field and deepen our understanding 🔬
Read more 👉 ndm.ox.ac.uk/news/study-on-t…

2
224

Biochemist Ivan Matić from @MPIAGE (Cologne) will present his research group's latest discoveries and developments on protein modification ADP-ribolysis at FLI on July 23. Find out more👉Link
#agingresearch #proteomics #proteinmodification #ADPribolisation #healthyaging
1
214
Biochemist Ivan Matić from @MPIAGE will present his research group's latest discoveries and developments on protein modification ADP-ribolysis at FLI on July 23. Find out more👉leibniz-fli.de/fli-colloquiu…
#agingresearch #proteomics #proteinmodification #ADPribolisation #healthyaging
2
198
Biochemist Ivan Matić from @MPIAGE will present his research group's latest discoveries and developments on protein modification ADP-ribolysis at FLI on July 23. Find out more 👉leibniz-fli.de/news-events/d…
#agingresearch #proteinmodification #ADPribolisation #healthyaging

ALT Ivan Matic (picture: private)
2
3
784

9 Jun 2024
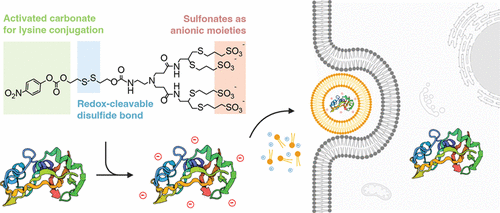
A modifier with multiple sulfonic acids and carbonate groups can alter protein surface charge and detach within target cells. An improved LNP version with fascinating potential applications. #LNP #ProteinModification
pubs.acs.org/doi/10.1021/acs…
3 Jun 2024

スルホン酸をたくさん繋ぎlinkerを挟んで、カーボネートを導入した修飾剤。これで修飾するとProteinの表面電荷を代えることが可能、かつTarget cell内で外れる仕様。LNPの改良版。コンセプトも面白いですが、この分子が興味深い。色々応用できそう。ACS Central Science
pubs.acs.org/doi/10.1021/acs…
1
5
683

18 Apr 2024
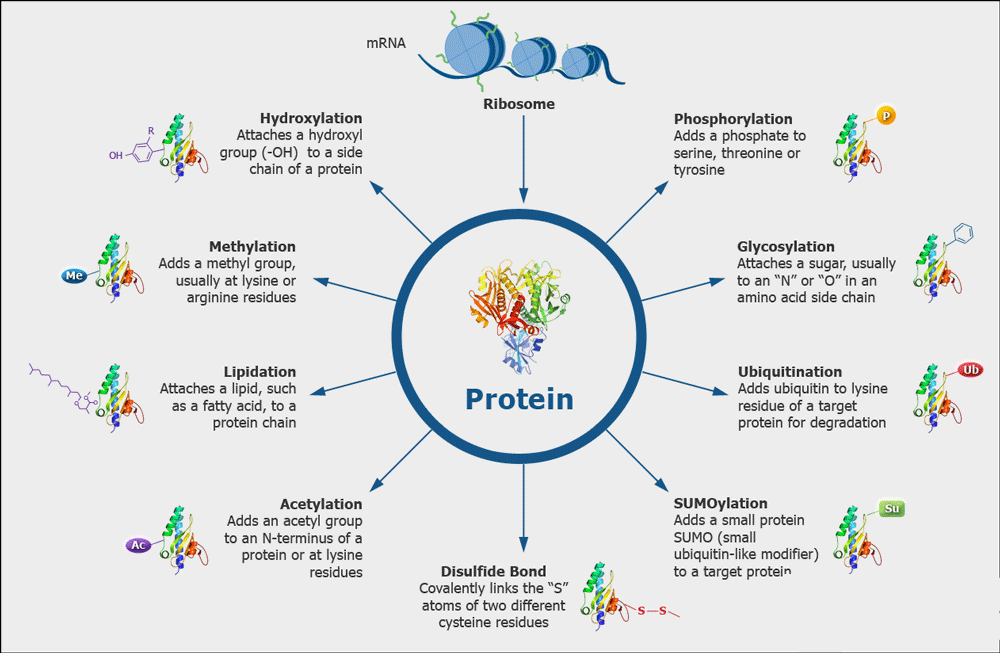
Misfolding, aggregation, variable glycosylation, oxidation of methionine, deamination of asparagine and glutamine, and proteolysis are common types of protein modifications.
#protein #proteinmodification #drugdevelopment
Image from creative proteomics
2
202

24 Nov 2023
Happy to share our recently online CHEM review about the precision in protein chemical modification and total synthesis🥳 Hit the link below to have a full view of it. authors.elsevier.com/c/1i8PB…
#ProteinModification #ProteinSynthesis cell.com/chem/fulltext/S2451…
3
11
1,135

2 Jul 2023
SUMOylation: A mechanism that modifies proteins by attaching a small protein called SUMO. It regulates protein activity, protein interactions, and various cellular processes. (10/19) #SUMOylation #ProteinModification #CellularProcesses
1
2
142

9 Nov 2022
Today in the CU dietetics kitchen, first years have been practicing protein modification. #dietetics #RD2B #proteinmodification #healthsciences


2
12

27 Oct 2022
📢 New Special Issue Open for Submissions: "Functional Properties of Food Proteins"
✏️ Guest edited by: Dr. Lina Zhang and Prof. Dr. Yulong Bao
mdpi.com/journal/molecules/s…
📌#proteinchemistry #proteinphysics #proteinmodification #proteomictechniques #foodmatrix
@ChemMatSci_MDPI

1
1

ADDR 50 days' free access:Expanding the chemical repertoire of protein-based polymers for drug-delivery applications.
By Osher Gueta & Miriam Amiram @bengurionu
#selfassembly #proteinpolymer #proteinmodification
authors.elsevier.com/a/1fokq…
1
2
5 Aug 2022
#RecommendedPaper #InterestingPaper
Halomethyl-Triazoles for Rapid, Site-Selective Protein Modification
By: Alison N. Hulme, et al.
👉mdpi.com/1420-3049/26/18/546…
📌#PTMs #halomethyl #triazole #proteinmodification
3

3 Feb 2021
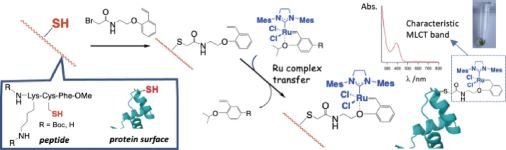
Ligand exchange | Ru-olefin interaction | Protein modification
Article by Dr. Takashi Matsuo @TakashiMatsuo6 @NAIST_MAIN_EN (NAIST) is available as Open Access
#LigandExchange #ProteinModification #Bioorthogonal #ChemicalModification
journal.csj.jp/doi/abs/10.12…
3
4

27 Jul 2020
Congratulations to Lee and colleagues who published their work in Molecular Cell using GeneTex’s ZHX2 antibody!👨🔬🔬👩🔬
“Proline Hydroxylation Primes Protein Kinases for Autophosphorylation and Activation”
#ProteinModification #citedantibodies #research


1
2

2 Aug 2019
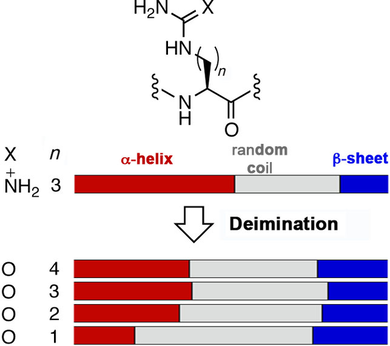
#Posttranslational #citrullination of arginine: effects of side-chain length on peptide secondary structure (Cheng at National Taiwan University) #proteinmodification doi.wiley.com/10.1002/cbic.2…
2
4