
Jun 11
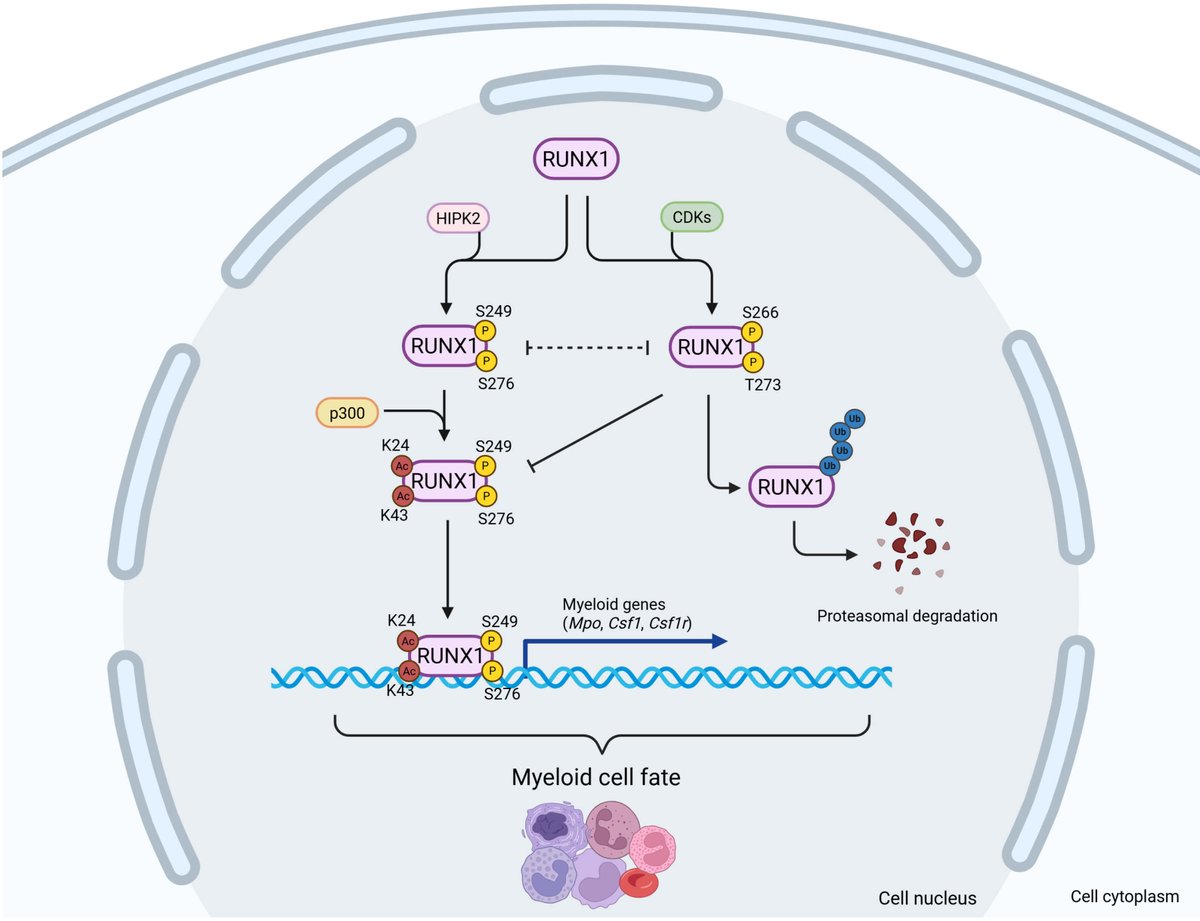
3/4 We identify novel substrates like #RPL22, #EIF3D and #PCM1, opening new doors to more functional insights into these #posttranslationalmodifications so far implicated predominantly in #microtubule regulation. Their role in #cellbiology seems more than what is thought to be.
1
1
156

This review highlights how #PostTranslationalModifications coordinate #InfluenzaVirus infection—from entry and replication to immune evasion—revealing interconnected regulatory networks and new opportunities for #antiviral and #vaccine development.
Read: doi.org/10.1016/j.gendis.202…
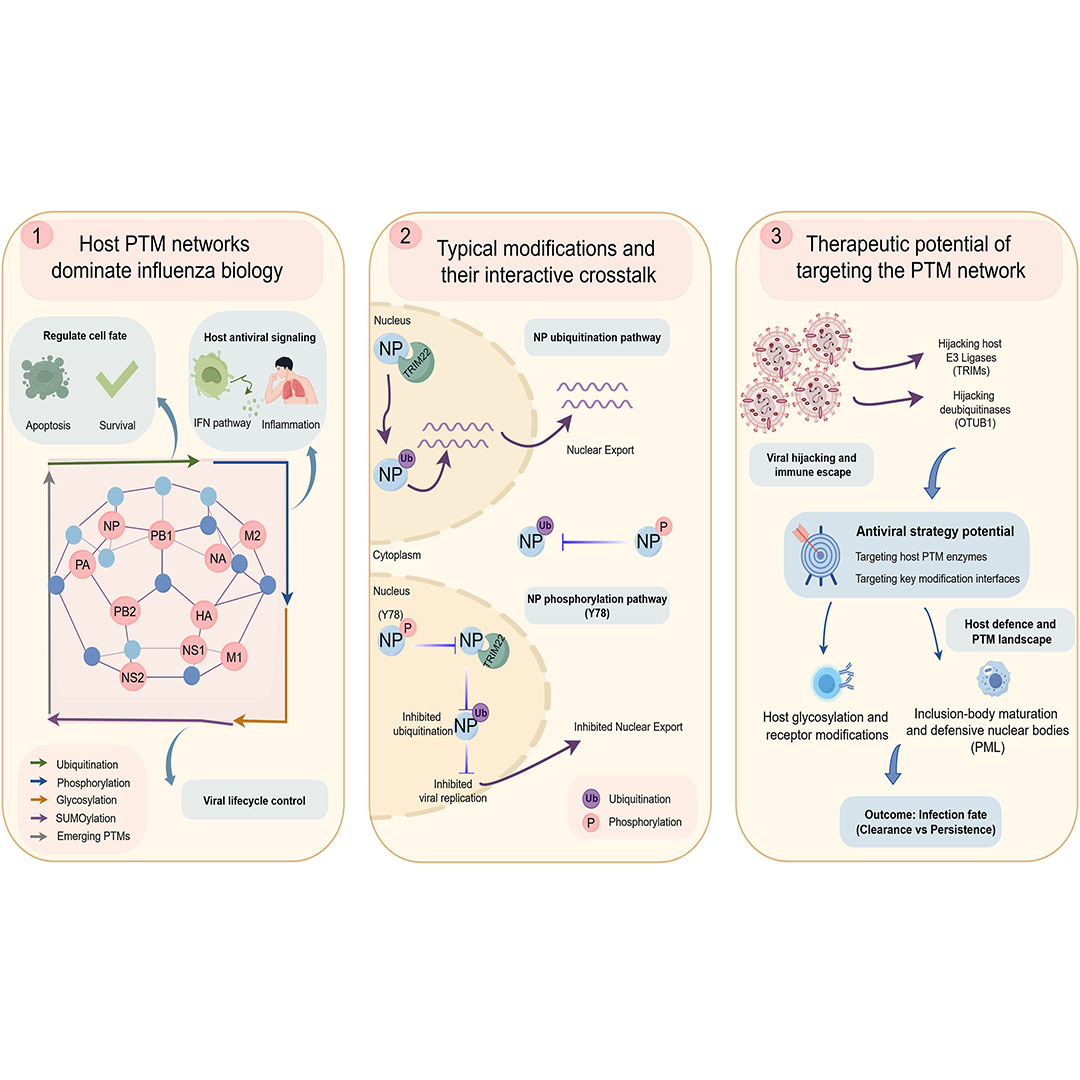
1
52
Learning the PTM Code through a Coarse-to-Fine Mechanism-Aware Framework
1. The paper introduces COMPASS-PTM, a two-stage framework that jointly learns residue-level multi-label PTM site profiles and enzyme–substrate assignment, framing them as two resolutions (coarse-to-fine) of the same underlying PTM “regulatory program.”
2. Stage 1 (MSPN) targets realistic PTM complexity: multiple PTM types can co-occur at the same residue, and predictions should remain biologically coherent rather than treating each PTM/site independently.
3. A key idea is crosstalk-aware prompting: a learnable PTM-type × PTM-type prior matrix (initialized from empirical PTM co-occurrence) is injected as an attention bias, encouraging predictions that reflect cooperative/antagonistic PTM relationships and reducing implausible multi-label outputs.
4. MSPN uses a dual-modal encoder: a protein language model backbone (ESM2-150M) for evolutionary/sequence context plus a chemical modality (SELFIES-based chemical language model embeddings physicochemical descriptors) to capture residue reactivity; fusion is explicitly designed to keep PLM features primary while selectively augmenting with chemical signals.
5. The training objective explicitly addresses PTM’s “dual long-tail” imbalance: rare PTM types across classes and extreme sparsity of modified residues across positions. A hybrid loss (macro regularized Dice micro focal, with additional regularization against over-confidence) is used to improve rare-class learning while controlling false positives.
6. On multi-label proteome-scale benchmarks curated from dbPTM and qPTM, MSPN reports large gains over evaluated baselines, including a 122% relative improvement in macro-F1 for multi-label site prediction; the paper emphasizes that improvements are driven by better precision (practical for experimental follow-up), not just AUROC.
7. Beyond per-residue metrics, COMPASS-PTM reduces “spurious multi-label burden” (over-calling extra PTM types once a site is detected), producing cleaner PTM label sets and higher protein-level “program” coherence.
8. Stage 2 (ESPS) reuses the PTM-aware substrate representation from Stage 1 and refines it into enzyme-resolved hypotheses by learning enzyme embeddings and predicting enzyme–substrate relationships; evaluation includes warm-start and stringent cold-start splits for unseen substrates or unseen enzymes.
9. ESPS improves enzyme assignment on OmniPath and SAGEPhos benchmarks, and achieves strong generalization in a true zero-shot setting on DARKIN (disjoint kinases between train/test), reporting a 54% mAP gain over the previously best reported score.
10. The framework is positioned as interpretable and variant-aware: embeddings organize into biochemically meaningful clusters; recovered kinase-family motifs match canonical consensus patterns; and case studies link missense variants to both local PTM gain/loss and predicted rewiring of enzyme–substrate interactions (e.g., LRRK2 variants affecting PKA-linked phosphorylation; hypotheses for SCNN1B and FUS).
💻Code: github.com/ZhangJJ26/COMPASS…
📜Paper: doi.org/10.1038/s41467-026-7…
#ComputationalBiology #Bioinformatics #Proteomics #PostTranslationalModifications #ProteinLanguageModels #DeepLearning #SystemsBiology #Kinase #Ubiquitination #VariantInterpretation
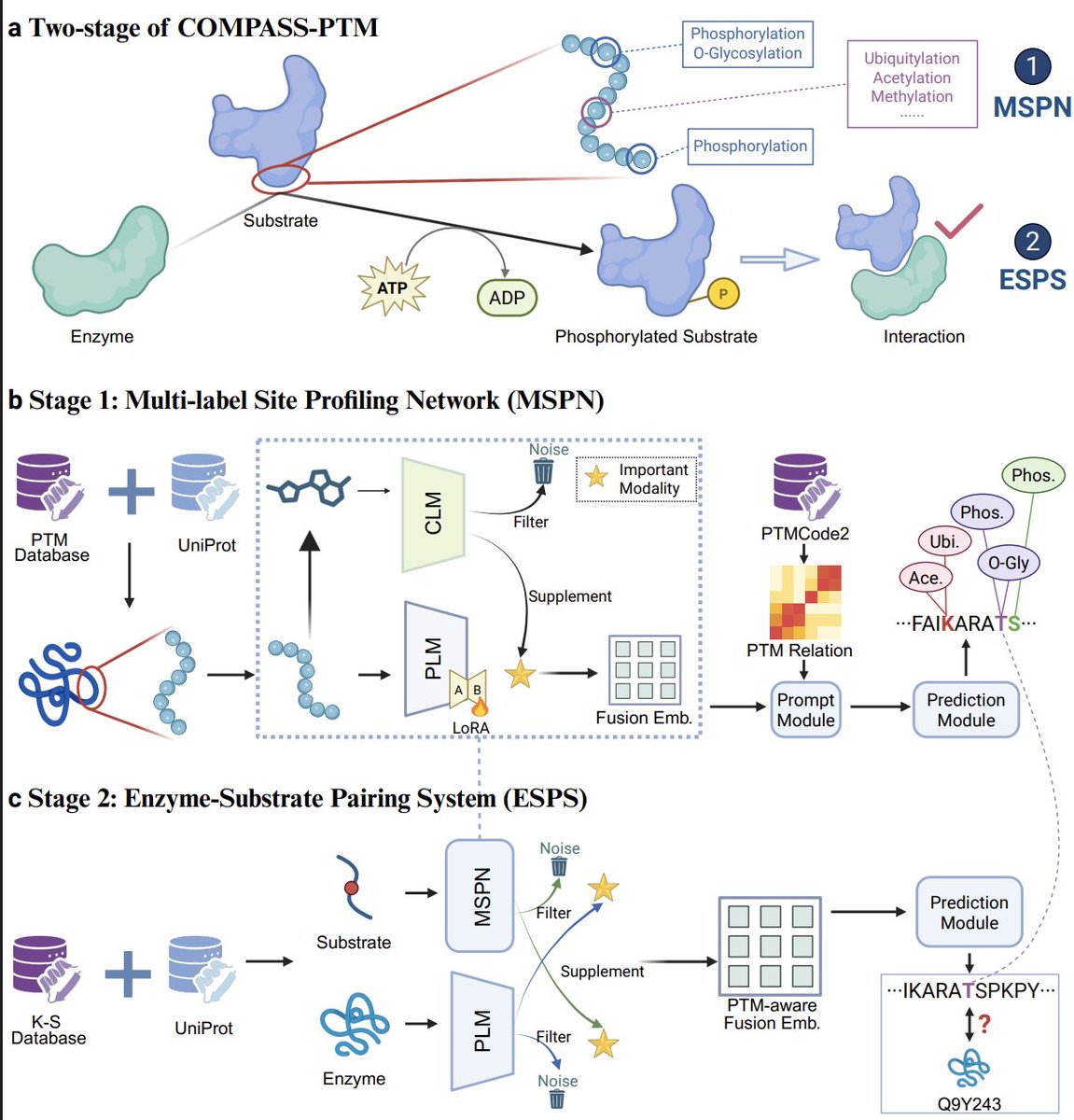
5
25
2,414
An LLM-driven pipeline for proteomics-based detection and structural modeling of post-translational modifications
1. The paper presents an integrated, LLM-driven workflow that connects open-search MS proteomics PTM discovery to downstream structural/dynamic modeling, aiming to turn “delta-mass lists” into mechanistic hypotheses about how PTMs regulate proteins.
2. The pipeline has two coupled components: PTMdiscoverer (LLM-assisted PTM identification/annotation from open-search results) and PTM-Psi (structural modeling of PTM effects on protein conformations, dynamics, and interactions), bridging detection with structural interpretation.
3. PTMdiscoverer starts from MSFragger open-search outputs, then applies a multi-stage quality localization cascade (e.g., stringent PSM probability, missed-cleavage filters, and localization confidence constraints) to produce high-confidence candidate modification events before any LLM reasoning.
4. The key addition vs conventional PTM summarization is LLM-based contextual prioritization: for each protein, a structured zero-shot prompt provides experimental context (e.g., TMT labeling, NEM thiol blocking, organism/conditions) and asks the model to map delta masses to PTM types (within tolerance), annotate residue positions, and propose functional relevance in a controlled vocabulary JSON output.
5. Case study: cyanobacterial “dark complex” proteins (GAPDH/GAP2, CP12, PRK) from Synechococcus elongatus under light disturbance. After filtering, the candidate event counts were large (CP12 92, PRK 394, GAP2 171), motivating automated prioritization.
6. The prioritized PTMs were dominated by cysteine-centered redox chemistry. Across all three proteins, 15.994 to 15.997 Da on cysteine was interpreted as oxidation consistent with sulfenylation (Cys-SOH), aligning with known redox regulation of dark complex assembly and Calvin-cycle control.
7. PTMdiscoverer also flagged NEM alkylation signatures (e.g., ~57.029 Da and ~125.044 Da) as sample-prep/thiol-blocking artifacts rather than endogenous PTMs—an example of using experimental context to avoid misinterpreting chemistry introduced by the workflow.
8. The structural modeling stage (PTM-Psi) is positioned to take residue-resolved PTM annotations and predicted structures (e.g., AlphaFold-like inputs) to simulate PTM-dependent conformational/dynamic changes and interaction effects, enabling hypothesis generation about how redox-linked PTMs tune enzyme states and complex formation.
9. Engineering/reproducibility: PTMdiscoverer is provided as a Python package with CLI plus an MCP-compatible server exposing tools for validation, protein listing, delta-mass extraction, inference, and deterministic/multi-run consensus analysis; a containerized Streamlit app (via ADEPT agentic orchestration) integrates auxiliary tools (sequence/chemical DB queries, RAG) for interactive analysis.
10. Limitations noted: LLM non-determinism (addressed via multi-run consensus tooling), dependence on a commercial API in the presented runs (but configurable for other endpoints/models), and limited biological breadth in this preprint (3 proteins, one organism), motivating future benchmarking and tighter end-to-end automation into PTM-Psi perturbation studies.
💻Code: github.com/pnnl/PTMdiscovere…
📜Paper: biorxiv.org/content/10.64898…
#Proteomics #MassSpectrometry #PostTranslationalModifications #PTM #LLM #GenerativeAI #ComputationalBiology #StructuralBiology #ProteinDynamics #Cyanobacteria
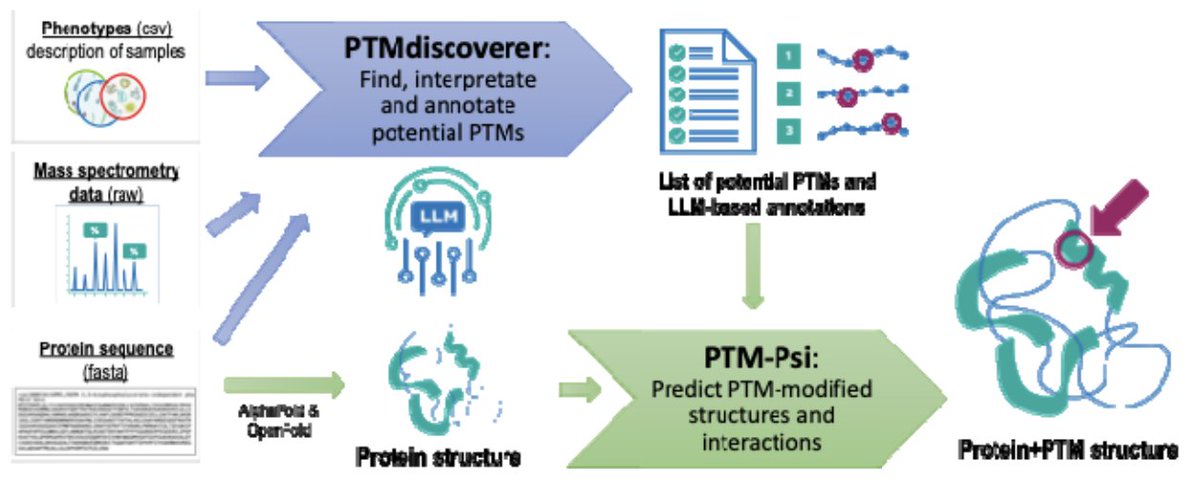
3
19
1,326

The latest work from Institut National de la Recherche Scientifique @inrsciences by first author Dr. Pegah Ghavidel and her team is now featured in FBL!
Welcome to read this article 👉: imrpress.com/journal/FBL/31/…
Article Highlights
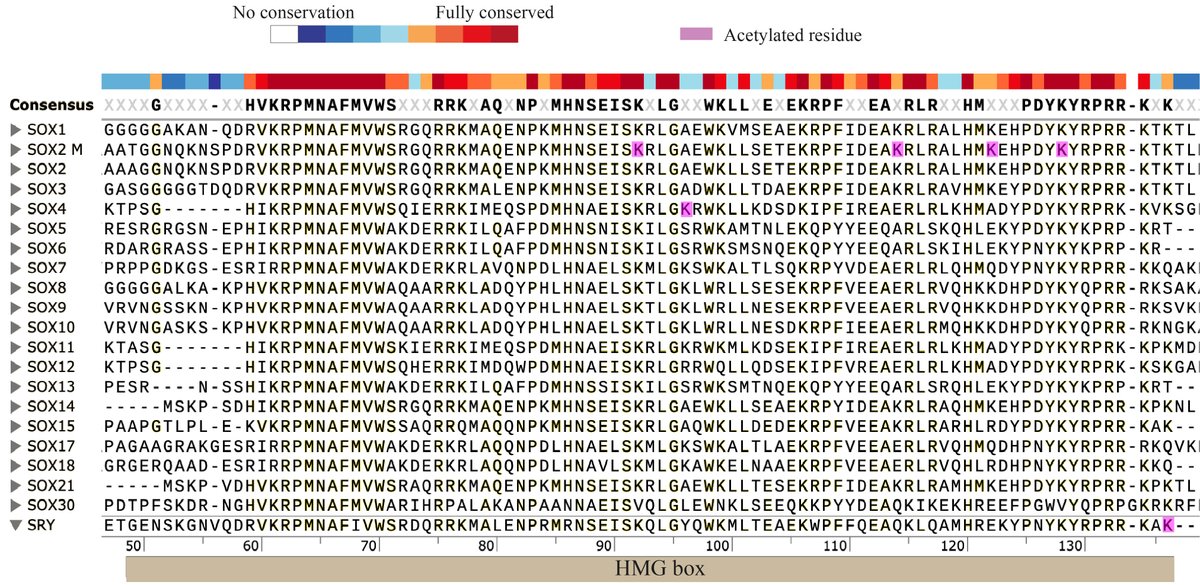
💡Is transcription factor acetylation the missing link between epigenetic regulation and cell fate decisions?
This review highlights how direct acetylation of transcription factors acts as a precise, context-dependent molecular switch shaping lineage specification, cellular plasticity, and therapeutic potential.
✒️ Author: Pegah Ghavidel, Yassine Abdelmalki, Marie-Claude Sincennes @MarieSincennes (Corresponding author)
🏪 Affiliation: Institut National de la Recherche Scientifique @inrsciences ; Unité Mixte de Recherche INRS-UQAC en Santé Durable
📖 Don’t miss this citable reference for your future research!
Citation format: Ghavidel, Pegah, Yassine Abdelmalki, and Marie-Claude Sincennes. "Transcription Factor Acetylation and Cell Fate Control: A Molecular Switch in Hematopoiesis and Myogenesis." Frontiers in Bioscience-Landmark 31.3 (2026).
Welcome to submit your work via our submission link👉: imr.propub.com/
FBL is dedicated to the publication of original articles, reviews, methods and commentaries in all areas of cellular and molecular biology.
📖 Click here for author guidelines: imrpress.com/journal/FBL/pag…
#Epigenetics #TranscriptionFactors #CellFate #StemCells #Hematopoiesis #Myogenesis #PostTranslationalModifications #PrecisionMedicine #FBLPaper

1
7
1,134

Feb 23
Planning to attend @agbt? Now is the time to learn about the latest in next-gen protein sequencing technology from Quantum-Si. Head of Scientific Affairs, Meredith Carpenter, PhD, will be sharing how to achieve high resolution #PTM data.
Find her at poster 407 on Tuesday to learn more.
#QSI #AGBT2026 #PostTranslationalModifications #PTMAnaylsis #ProteinCharacterization #NGPS #Proteomics
4
2
6
981
Feb 20
#USHUPO we will see you next week! On Tuesday, an exciting opportunity to see the data out of the lab of one of our customers, Dr. @GSheynkman from @UVA.
Drop by her presentation on successful #PTM detection with Quantum-Si's protein sequencing and stay to get a sneak peek at our expanding capabilities.
#QSI #USHUPO26 #PostTranslationalModifications #ProteinAnalysis #Proteomics #Multiomics #ProteinSequencing

2
1
4
645

14 Oct 2025
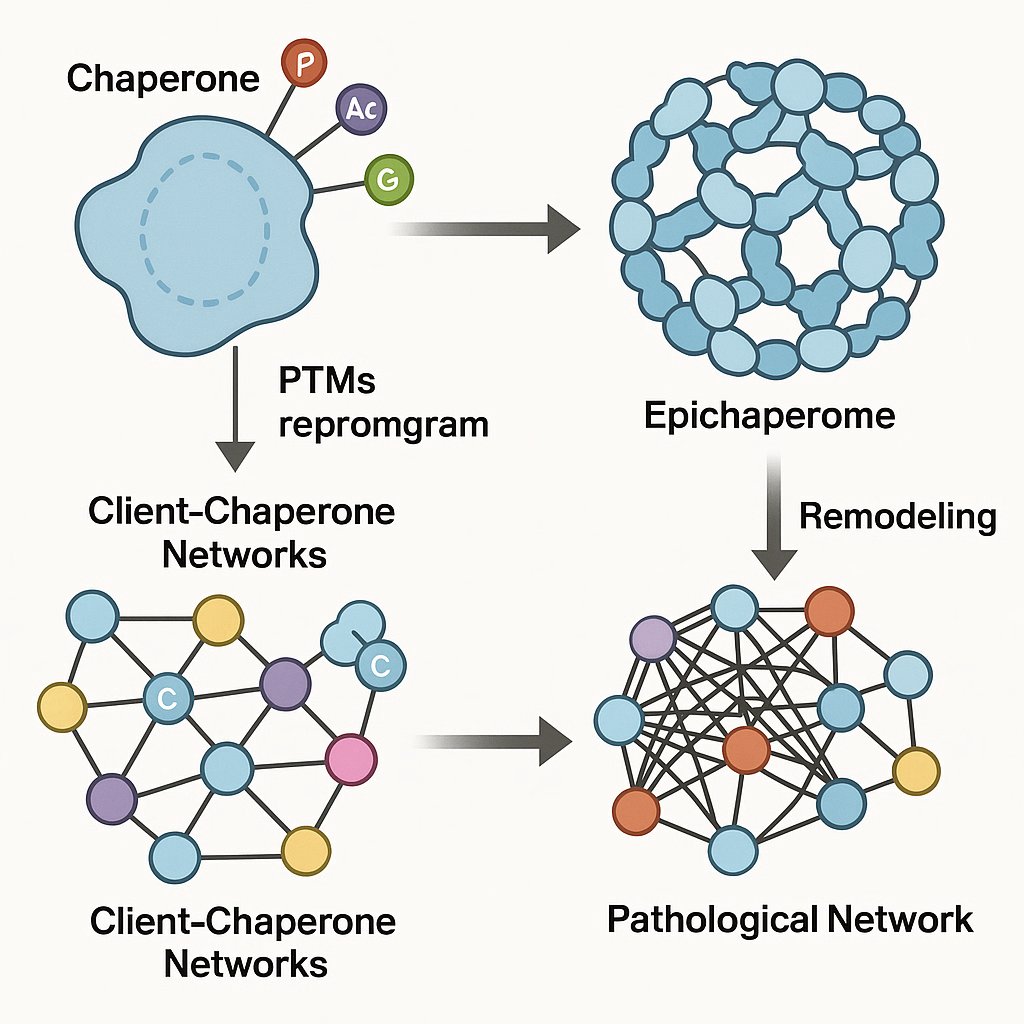
In our recent @TrendsBiochem review, we propose that #PTMs do far more than modulate single-protein behavior.
Further explore how #posttranslationalmodifications can rewrite cellular networks➡️@SpringerNature Communities: go.nature.com/4o8jGQD
#epichaperome #networkbiology
4
8
631

25 Jun 2025
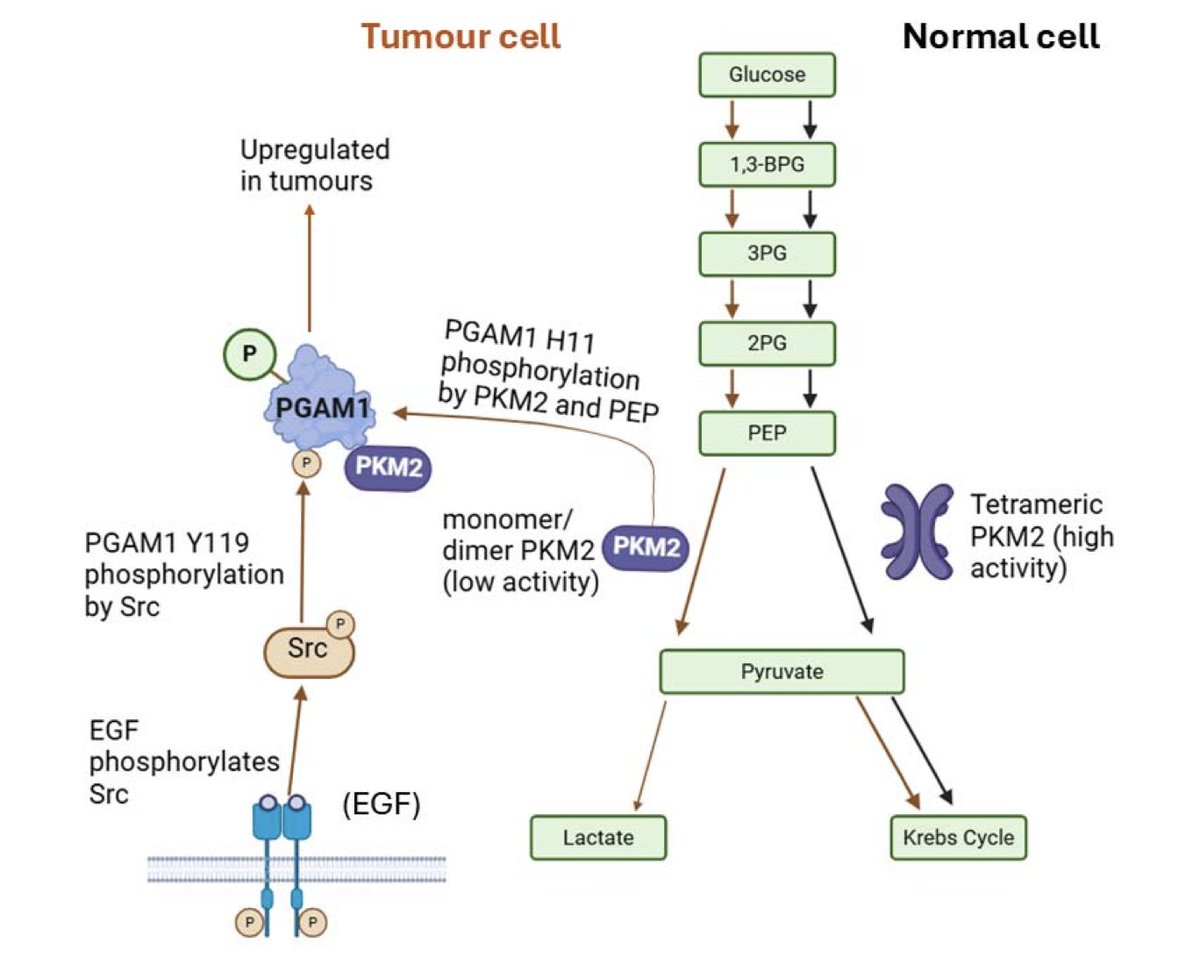
AlphaFold 3-enabled in silico exploration of PGAM1 interactions in cancer
1.This study leverages AlphaFold 3 to explore how post-translational phosphorylation at tyrosine 119 (Y119) in PGAM1 enables access to its catalytic site, explaining a non-canonical activation route relevant to cancer metabolism.
2.Phosphorylation at Y119 causes a large conformational rearrangement in PGAM1's C-terminal tail, opening up the catalytic H11 site to allow binding by phosphoenolpyruvate (PEP)—a key step for PGAM1 activation via PKM2.
3.In AF3 models, the movement of Lys254 by over 37 Å following Y119 phosphorylation unblocks the H11 site, supporting a structural mechanism for enhanced enzymatic activity via PEP binding.
4.Ligand docking using Webina and SwissDock showed that pY119-PGAM1 models consistently enable more frequent and catalytically relevant binding of PEP near H11, unlike unphosphorylated structures.
5.DiffDock did not show a significant difference in PEP binding between phosphorylated and unphosphorylated forms—likely due to its lack of binding site specification—highlighting the importance of tool choice in docking studies.
6.Despite Y119 phosphorylation enabling PEP access, structural modelling failed to produce PGAM1–PKM2 complexes with proximal catalytic sites. Neither AF3 nor ClusPro identified feasible configurations, suggesting missing dynamic elements.
7.These findings suggest PGAM1 activation by PKM2 may not rely on direct catalytic site alignment but rather on a broader conformational priming effect—potentially involving transient or multi-step assembly processes.
8.This work illustrates how phosphorylation-driven allosteric regulation can rewire enzyme accessibility in cancer, reinforcing the Warburg effect by enabling PGAM1 to accelerate glycolysis under oncogenic conditions.
9.The unusually large conformational changes predicted by AF3 (if validated experimentally) could represent an extreme example of PTM-driven structural plasticity, offering new insights into regulatory flexibility of cancer-associated enzymes.
10.By combining AlphaFold 3, small molecule docking, and protein-protein interaction modelling, this study presents a blueprint for mechanistic investigations of PTM-controlled protein function in disease contexts.
📜Paper: biorxiv.org/content/10.1101/…
#AlphaFold3 #PGAM1 #CancerMetabolism #PostTranslationalModifications #StructuralBiology #ComputationalBiology #Glycolysis #WarburgEffect
2
20
1,291

18 Jun 2025
How iron helps collagen enzymes work - and what happens when it doesn’t @MedUniGraz @ActaCrystF @IUCr #PostTranslationalModifications #DNAViruses #Dioxygenases doi.org/10.1107/S2053230X250…
3
131
7 May 2025
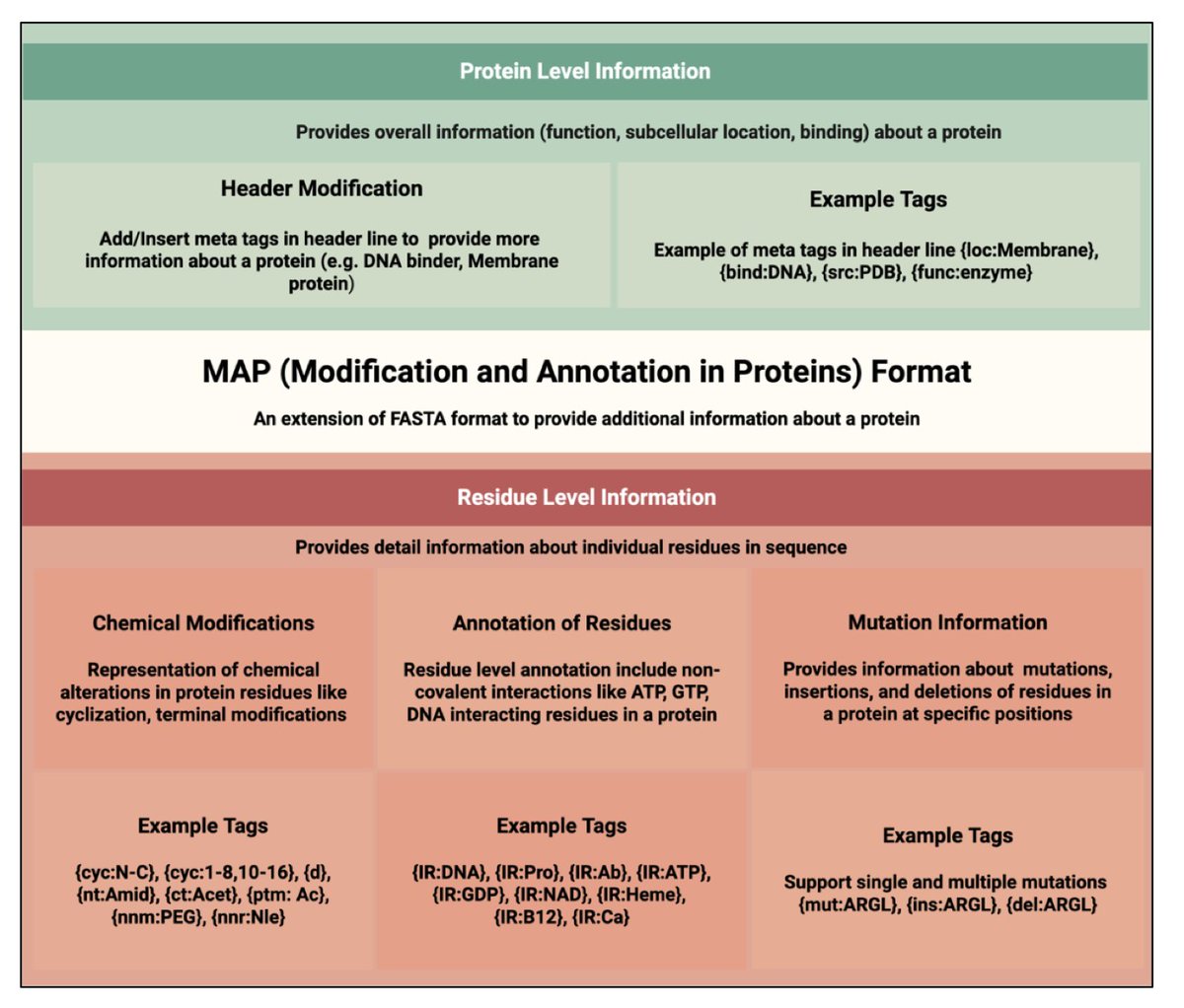
MAP Format for Representing Chemical Modifications, Annotations, and Mutations in Protein Sequences: An Extension of the FASTA Format
1. This study proposes MAP, a new sequence format that extends the widely used FASTA format to include in-line annotations for protein modifications, mutations, and structural features—retaining readability and compatibility with existing bioinformatics tools.
2. MAP uses intuitive curly-brace tags embedded directly within the protein sequence to denote modifications like phosphorylation, cyclization, non-natural residues, or D-amino acids, providing residue-level annotation in a single-line, human-readable format.
3. Header metadata is structured using tags like {org\:Homo sapiens} or {func\:DNA-binding}, allowing easy parsing of protein-level attributes like organism, subcellular location, and function—enhancing integration with protein databases.
4. Unlike PEFF, HELM, or BILN, which are either too simplistic or overly complex, MAP strikes a balance—simple enough for biologists to edit manually, yet expressive enough to represent therapeutic peptides and engineered proteins with chemical modifications.
5. MAP can denote sequence variants such as mutations (e.g., {mut\:R}), insertions ({ins\:MK}), and deletions ({del\:RTH}) within the linear sequence, enabling unified representation of wild-type and mutant forms for variant annotation and analysis.
6. The format supports tagging of interacting residues using {IR\:Target} (e.g., H{IR\:Zn}), enabling capture of functional sites like metal ion binding or nucleic acid interaction sites directly within the sequence.
7. Applications span protein therapeutics, synthetic biology, structural bioinformatics, and database curation. MAP is already positioned to enhance resources like UniProt, PDB, and peptide therapeutic databases by embedding rich residue-level context.
8. MAP is backward-compatible: removing all curly-brace tags produces a valid FASTA sequence, ensuring that legacy tools can still interpret core sequence content while MAP-aware tools leverage annotations.
9. The authors provide a web portal (MAPrepo) offering manuals, curated datasets, database exports, and Python scripts for converting between formats, facilitating immediate adoption by the community.
10. MAP’s simplicity, flexibility, and compatibility position it as a practical standard for next-generation protein sequence annotation—bridging raw sequences and chemically informed representations in a unified, FAIR-compliant format.
💻Code: webs.iiitd.edu.in/raghava/ma…
📜Paper: arxiv.org/abs/2505.03403
#ProteinSequence #FASTA #MAPformat #Bioinformatics #ProteinModification #PostTranslationalModifications #DataStandards #Proteomics #ComputationalBiology #SequenceAnnotation #TherapeuticProteins #FAIRdata

1
2
670
7 May 2025
MAP Format for Representing Chemical Modifications, Annotations, and Mutations in Protein Sequences: An Extension of the FASTA Format
1. This study proposes MAP, a new sequence format that extends the widely used FASTA format to include in-line annotations for protein modifications, mutations, and structural features—retaining readability and compatibility with existing bioinformatics tools.
2. MAP uses intuitive curly-brace tags embedded directly within the protein sequence to denote modifications like phosphorylation, cyclization, non-natural residues, or D-amino acids, providing residue-level annotation in a single-line, human-readable format.
3. Header metadata is structured using tags like {org\:Homo sapiens} or {func\:DNA-binding}, allowing easy parsing of protein-level attributes like organism, subcellular location, and function—enhancing integration with protein databases.
4. Unlike PEFF, HELM, or BILN, which are either too simplistic or overly complex, MAP strikes a balance—simple enough for biologists to edit manually, yet expressive enough to represent therapeutic peptides and engineered proteins with chemical modifications.
5. MAP can denote sequence variants such as mutations (e.g., {mut\:R}), insertions ({ins\:MK}), and deletions ({del\:RTH}) within the linear sequence, enabling unified representation of wild-type and mutant forms for variant annotation and analysis.
6. The format supports tagging of interacting residues using {IR\:Target} (e.g., H{IR\:Zn}), enabling capture of functional sites like metal ion binding or nucleic acid interaction sites directly within the sequence.
7. Applications span protein therapeutics, synthetic biology, structural bioinformatics, and database curation. MAP is already positioned to enhance resources like UniProt, PDB, and peptide therapeutic databases by embedding rich residue-level context.
8. MAP is backward-compatible: removing all curly-brace tags produces a valid FASTA sequence, ensuring that legacy tools can still interpret core sequence content while MAP-aware tools leverage annotations.
9. The authors provide a web portal (MAPrepo) offering manuals, curated datasets, database exports, and Python scripts for converting between formats, facilitating immediate adoption by the community.
10. MAP’s simplicity, flexibility, and compatibility position it as a practical standard for next-generation protein sequence annotation—bridging raw sequences and chemically informed representations in a unified, FAIR-compliant format.
💻Code: webs.iiitd.edu.in/raghava/ma…
📜Paper: arxiv.org/abs/2505.03403
#ProteinSequence #FASTA #MAPformat #Bioinformatics #ProteinModification #PostTranslationalModifications #DataStandards #Proteomics #ComputationalBiology #SequenceAnnotation #TherapeuticProteins #FAIRdata
2
549
3 May 2025
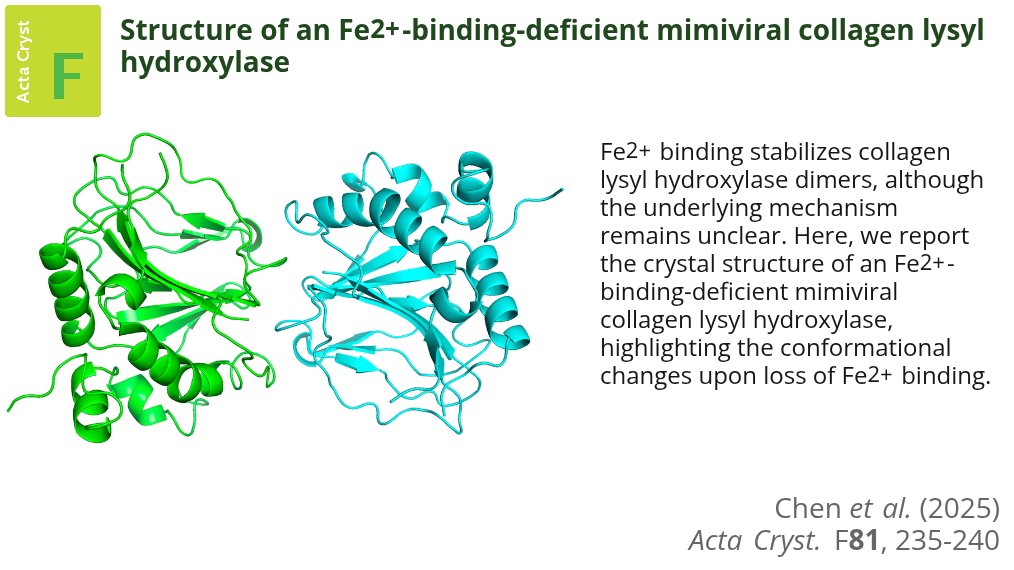
Tingfei Chen et al.: Structure of an Fe2 -binding-deficient mimiviral collagen lysyl hydroxylase #PostTranslationalModifications #DNAViruses #Dioxygenases @MedUniGraz... #IUCr journals.iucr.org/paper?S205…
1
169

2 May 2025
#DLSResearchSeminar 🍁
Join us today for a talk by Dr. Rashna Bhandari, Staff Scientist, CDFD, Hyderabad, on the importance of protein pyrophosphorylation✨
Title: A high-energy phosphate jump - from pyro-phosphoinositol to pyro-phosphoproteins.
#posttranslationalmodifications

8
256
19 Apr 2025
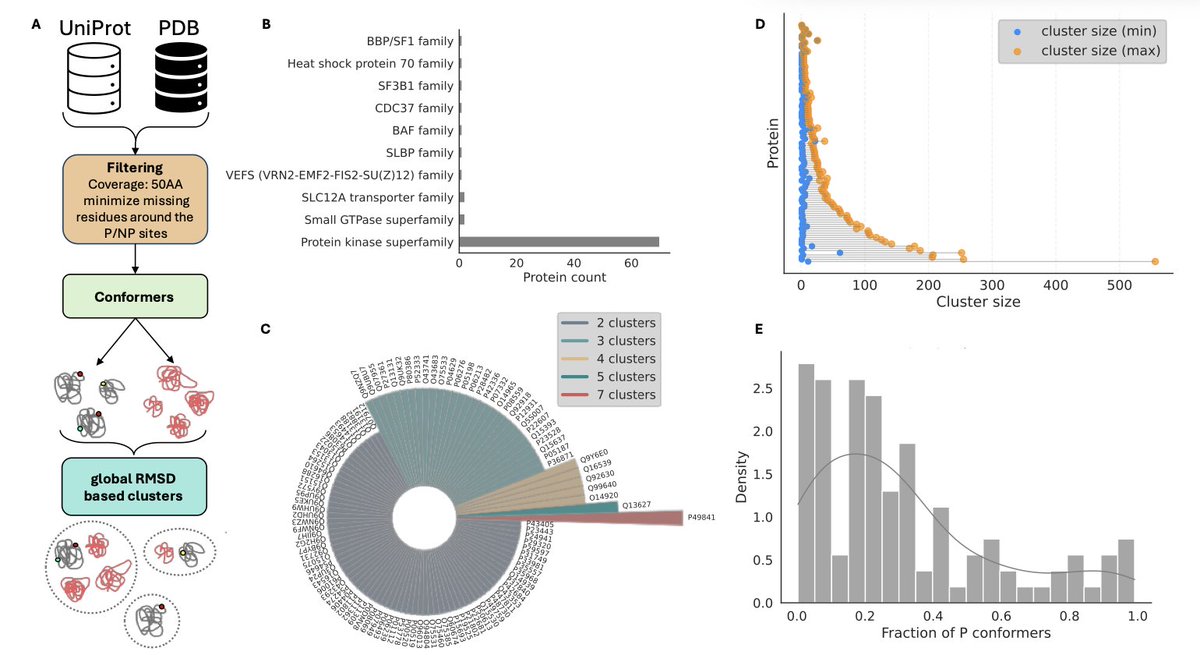
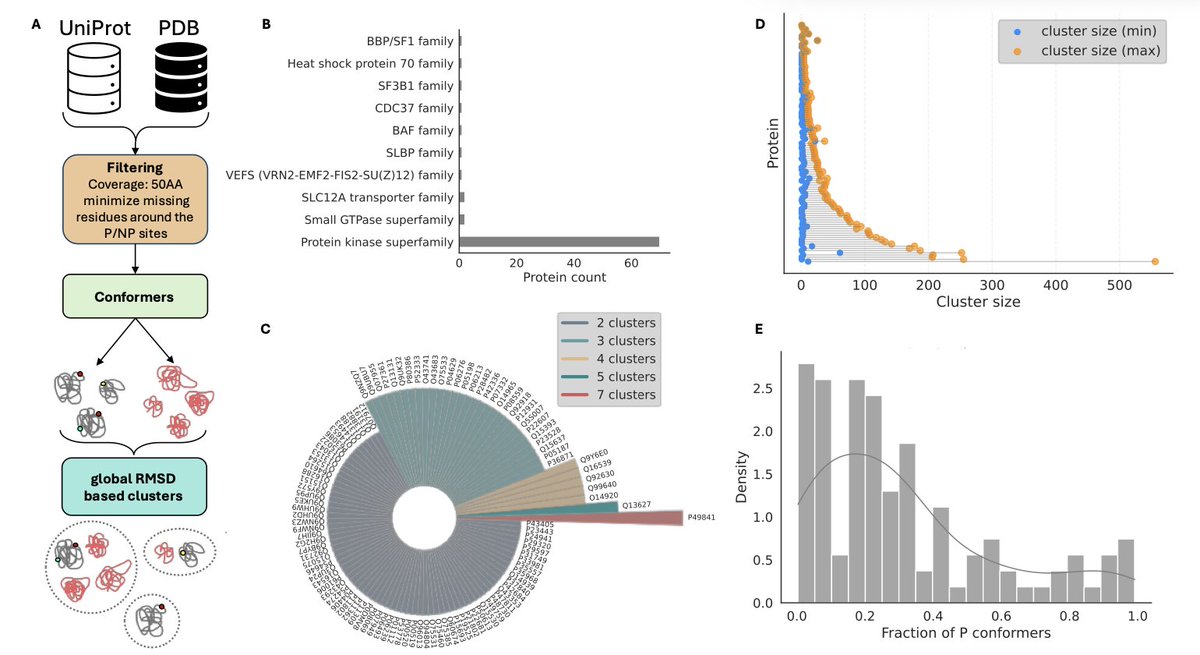
Assessing the relation between protein phosphorylation, AlphaFold3 models and conformational variability
1. This study systematically evaluates how well AlphaFold models capture phosphorylation-induced structural changes, using 109 proteins with experimentally determined phosphorylated (P) and non-phosphorylated (NP) conformers from the PDB.
2. Despite advances in AlphaFold3, all tested models—AF2, AF3, and AF3-p—predominantly predict the dominant conformational state, showing limited sensitivity to phosphorylation-driven structural diversity.
3. The study introduces conformational diversity scores (CDS and pCDS) to quantify structural variability. Only 23% of proteins showed clear structural shifts upon phosphorylation, with most phosphorylations subtly stabilizing pre-existing conformations.
4. Phosphorylation was found to fine-tune rather than radically alter protein structures, with 65% of clusters containing both P and NP conformers. This suggests that many phosphorylation effects are regulatory rather than structurally transformative.
5. Single-site phosphorylation events, especially involving phosphotyrosine (PTR), introduced the most pronounced local and global conformational shifts, while phosphoserine (SEP) had minimal impact, consistent with its stabilizing role.
6. AlphaFold3-p, the phosphorylation-aware model, showed only modest improvement over AF2 and AF3, aligning with phosphorylated states in 73% of cases versus 70% (AF3) and 64% (AF2), with no statistically significant differences.
7. All models consistently favored dominant structural clusters, likely due to biases in the PDB training data and co-evolutionary constraints, limiting their ability to explore rare or functionally relevant alternate states.
8. Structural predictions by AlphaFold models clustered together in over 80% of proteins, and all models aligned with the most dominant structural ensemble in 98% of cases, regardless of phosphorylation.
9. Residue-level analysis showed that phosphorylation-induced changes are more likely in structured domains and flexible coil regions with increased solvent accessibility, particularly in multi-domain proteins.
10. The study underscores a key limitation in current deep learning models: while excellent at predicting stable conformations, they struggle to model PTM-induced flexibility, which is often critical for biological function.
11. Authors advocate for richer training datasets with more diverse PTM-containing structures, and propose refining AlphaFold-like models to better capture local structural adjustments and rare phosphorylation-driven conformations.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinStructure #AlphaFold3 #Phosphorylation #PostTranslationalModifications #StructuralBiology #AI4Science #ComputationalBiology
5
1,170
19 Apr 2025
Assessing the relation between protein phosphorylation, AlphaFold3 models and conformational variability
1. This study systematically evaluates how well AlphaFold models capture phosphorylation-induced structural changes, using 109 proteins with experimentally determined phosphorylated (P) and non-phosphorylated (NP) conformers from the PDB.
2. Despite advances in AlphaFold3, all tested models—AF2, AF3, and AF3-p—predominantly predict the dominant conformational state, showing limited sensitivity to phosphorylation-driven structural diversity.
3. The study introduces conformational diversity scores (CDS and pCDS) to quantify structural variability. Only 23% of proteins showed clear structural shifts upon phosphorylation, with most phosphorylations subtly stabilizing pre-existing conformations.
4. Phosphorylation was found to fine-tune rather than radically alter protein structures, with 65% of clusters containing both P and NP conformers. This suggests that many phosphorylation effects are regulatory rather than structurally transformative.
5. Single-site phosphorylation events, especially involving phosphotyrosine (PTR), introduced the most pronounced local and global conformational shifts, while phosphoserine (SEP) had minimal impact, consistent with its stabilizing role.
6. AlphaFold3-p, the phosphorylation-aware model, showed only modest improvement over AF2 and AF3, aligning with phosphorylated states in 73% of cases versus 70% (AF3) and 64% (AF2), with no statistically significant differences.
7. All models consistently favored dominant structural clusters, likely due to biases in the PDB training data and co-evolutionary constraints, limiting their ability to explore rare or functionally relevant alternate states.
8. Structural predictions by AlphaFold models clustered together in over 80% of proteins, and all models aligned with the most dominant structural ensemble in 98% of cases, regardless of phosphorylation.
9. Residue-level analysis showed that phosphorylation-induced changes are more likely in structured domains and flexible coil regions with increased solvent accessibility, particularly in multi-domain proteins.
10. The study underscores a key limitation in current deep learning models: while excellent at predicting stable conformations, they struggle to model PTM-induced flexibility, which is often critical for biological function.
11. Authors advocate for richer training datasets with more diverse PTM-containing structures, and propose refining AlphaFold-like models to better capture local structural adjustments and rare phosphorylation-driven conformations.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinStructure #AlphaFold3 #Phosphorylation #PostTranslationalModifications #StructuralBiology #AI4Science #ComputationalBiology
5
15
1,458
10 Apr 2025
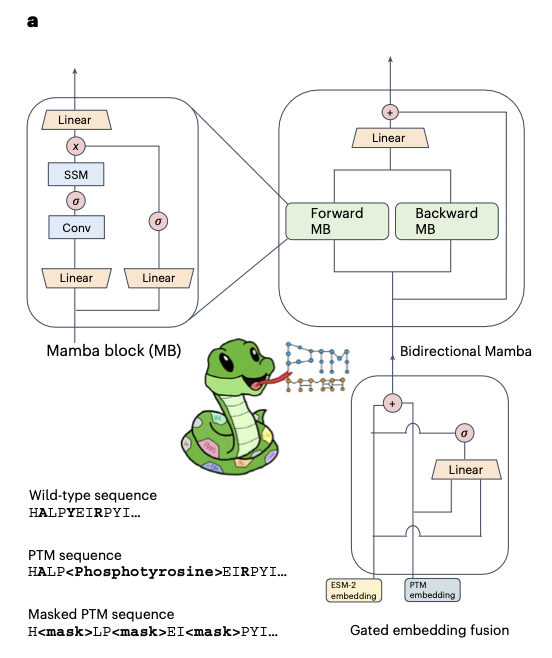
PTM-Mamba: a PTM-aware protein language model with bidirectional gated Mamba blocks @naturemethods
1. PTM-Mamba is the first protein language model explicitly designed to encode post-translational modifications (PTMs), using a novel bidirectional gated Mamba architecture fused with ESM-2 embeddings to represent both wild-type and modified residues.
2. The model introduces a new vocabulary of 27 PTM tokens, mapped from 311,000 experimentally validated records across 79,707 PTM-annotated sequences, enabling biologically grounded sequence modeling.
3. PTM-Mamba’s architecture incorporates forward and backward Mamba blocks with a gated fusion mechanism, combining ESM-2 and PTM-specific embeddings for rich, context-aware representations.
4. The model outperforms strong baselines like ESM-2, PTM-Transformer, and structure-aware PTM-SaProt in disease association, druggability prediction, and prediction of PTM-mediated effects on protein–protein interactions (PPIs).
5. On the PPI task using PTMint data, PTM-Mamba achieved the highest metrics in precision, F1, AUROC, and MCC, surpassing both structure-aware and sequence-based models in capturing PTM-dependent interaction dynamics.
6. PTM-Mamba supports zero-shot PTM discovery by predicting plausible PTMs from wild-type sequences without retraining, showing strong performance in identifying biologically relevant modifications like phosphoserine and diacylglycerol-cysteine.
7. Embedding analyses reveal that PTM-Mamba distinguishes PTM types via spatial clustering and preserves the contextual similarity of wild-type and modified pairs, validating its capacity to model PTM-specific functional effects.
8. The model maintains high performance in standard site-level PTM prediction tasks (e.g., phosphorylation, non-histone acetylation) while being optimized for sequence-level downstream applications.
9. PTM-Mamba was trained efficiently on masked language modeling with adaptive masking strategies, using an 8xA100 GPU system, and shows faster convergence than transformer-based baselines.
10. The model and its preprocessing toolkit are fully open-source and integrated into HuggingFace and GitHub, providing a scalable and modular platform for PTM-aware proteomics and therapeutic modeling.
💻Code: github.com/programmablebio/p…
📜Paper: nature.com/articles/s41592-0…
#ProteinDesign #PostTranslationalModifications #PTM #LLM #ProteinLanguageModel #Proteomics #Bioinformatics #ESM2 #Mamba #DrugDiscovery #PPI #PrecisionTherapeutics
1
13
1,391
10 Apr 2025
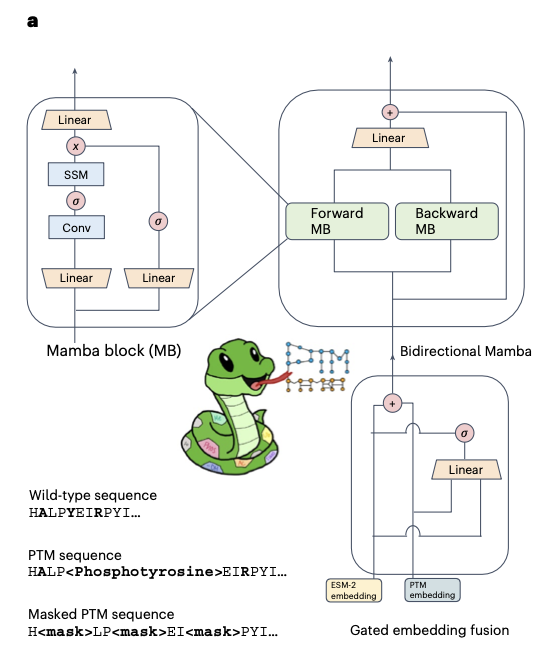
PTM-Mamba: a PTM-aware protein language model with bidirectional gated Mamba blocks @naturemethods
1. PTM-Mamba is the first protein language model explicitly designed to encode post-translational modifications (PTMs), using a novel bidirectional gated Mamba architecture fused with ESM-2 embeddings to represent both wild-type and modified residues.
2. The model introduces a new vocabulary of 27 PTM tokens, mapped from 311,000 experimentally validated records across 79,707 PTM-annotated sequences, enabling biologically grounded sequence modeling.
3. PTM-Mamba’s architecture incorporates forward and backward Mamba blocks with a gated fusion mechanism, combining ESM-2 and PTM-specific embeddings for rich, context-aware representations.
4. The model outperforms strong baselines like ESM-2, PTM-Transformer, and structure-aware PTM-SaProt in disease association, druggability prediction, and prediction of PTM-mediated effects on protein–protein interactions (PPIs).
5. On the PPI task using PTMint data, PTM-Mamba achieved the highest metrics in precision, F1, AUROC, and MCC, surpassing both structure-aware and sequence-based models in capturing PTM-dependent interaction dynamics.
6. PTM-Mamba supports zero-shot PTM discovery by predicting plausible PTMs from wild-type sequences without retraining, showing strong performance in identifying biologically relevant modifications like phosphoserine and diacylglycerol-cysteine.
7. Embedding analyses reveal that PTM-Mamba distinguishes PTM types via spatial clustering and preserves the contextual similarity of wild-type and modified pairs, validating its capacity to model PTM-specific functional effects.
8. The model maintains high performance in standard site-level PTM prediction tasks (e.g., phosphorylation, non-histone acetylation) while being optimized for sequence-level downstream applications.
9. PTM-Mamba was trained efficiently on masked language modeling with adaptive masking strategies, using an 8xA100 GPU system, and shows faster convergence than transformer-based baselines.
10. The model and its preprocessing toolkit are fully open-source and integrated into HuggingFace and GitHub, providing a scalable and modular platform for PTM-aware proteomics and therapeutic modeling.
💻Code: github.com/programmablebio/p…
📜Paper: nature.com/articles/s41592-0…
#ProteinDesign #PostTranslationalModifications #PTM #LLM #ProteinLanguageModel #Proteomics #Bioinformatics #ESM2 #Mamba #DrugDiscovery #PPI #PrecisionTherapeutics
5
15
1,190
8 Apr 2025
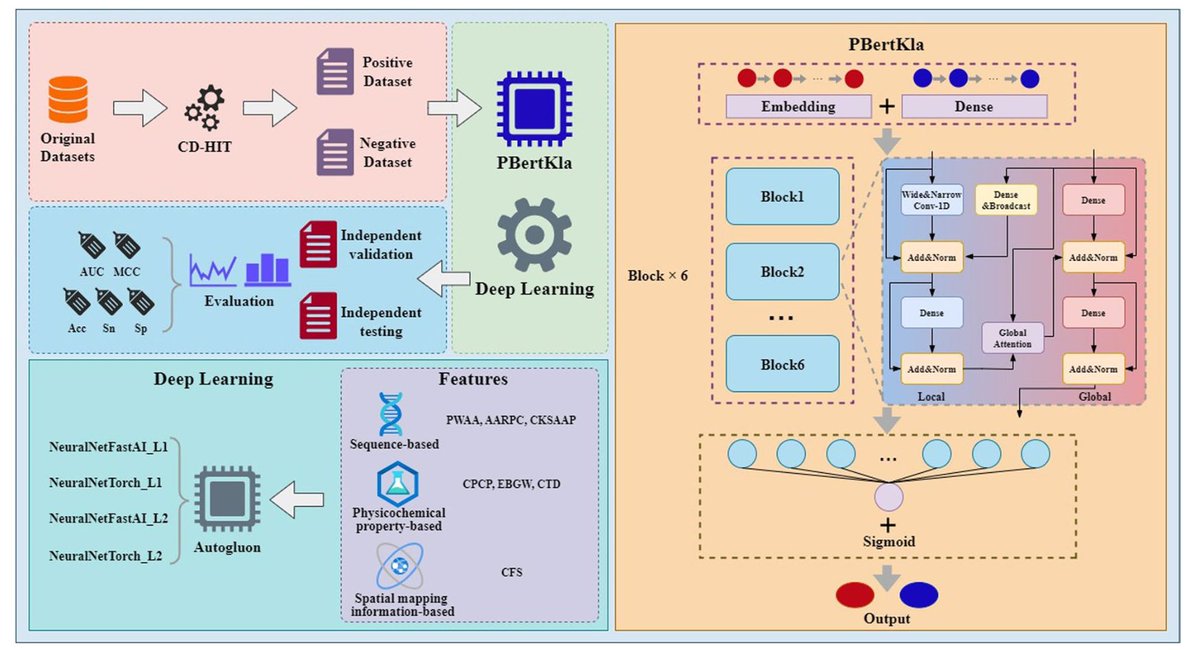
PBertKla: a protein large language model for predicting human lysine lactylation sites
1/ PBertKla is a protein language model tailored for predicting lysine lactylation (Kla) sites in human proteins. It leverages ProteinBERT, a deep learning model pretrained on protein sequences and Gene Ontology annotations, and fine-tunes it with Kla-specific data to capture both local and global sequence features.
2/ Lysine lactylation is a newly identified post-translational modification linked to diverse biological processes and diseases such as cancer and neurodegeneration. Accurately identifying Kla sites is critical for understanding these roles, but current experimental techniques are resource-intensive and computational tools are limited.
3/ The study first curated a high-quality Kla benchmark dataset, optimizing sequence window length (45 residues) and sequence identity threshold (30%) to ensure balanced and informative training data. This dataset underpins the model’s robust performance.
4/ PBertKla achieves an AUC of 0.884 and an AUPRC of 0.866 on independent validation sets, outperforming other state-of-the-art Kla prediction tools including DeepKla, AutoKla, and DeepKlapred. It also generalizes well to independent test sets derived from tumor and adjacent normal tissues.
5/ The model's dual-stream architecture processes both local (residue-level) and global (sequence-level) features through transformer-based blocks, with UMAP-based visualizations confirming progressively better Kla/non-Kla separation through the layers.
6/ Comparative analyses show that PBertKla exceeds the performance of handcrafted feature-based neural network models and other deep learning frameworks, even under rigorous conditions. It offers up to 9% improvements in accuracy and 19% in MCC over prior tools.
7/ Beyond prediction, PBertKla offers interpretability through feature visualization and its modular structure opens avenues for extending to Kla predictions in other species, incorporation of structural and evolutionary data, or integration into web-based analysis platforms.
8/ The paper highlights future directions such as building larger and more diverse Kla datasets, exploring multi-scale deep learning models like ESM3, and studying the interplay between Kla and other PTMs to deepen our understanding of protein regulation.
💻Code: github.com/laihongyan/PBertK…
📜Paper: bmcbiol.biomedcentral.com/ar…
#PostTranslationalModifications #ProteinLanguageModel #Bioinformatics #DeepLearning #Lactylation #Kla #ProteinBERT #ComputationalBiology #PTM #AIinBiology #Transformer #Epigenetics #Biomarkers
1
6
912
18 Mar 2025
Non-Canonical Crosslinks Confound Evolutionary Protein Structure Models
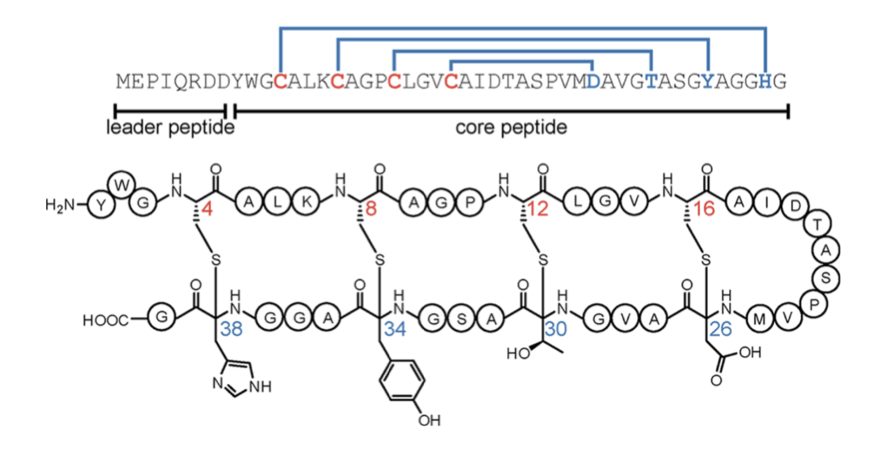
- Evolution-based protein structure prediction models like AlphaFold and RoseTTAFold have revolutionized structural biology but struggle with rare post-translational modifications (PTMs). This study evaluates their limitations in predicting sactipeptides, a class of ribosomally synthesized and post-translationally modified peptides (RiPPs) with unique sulfur-to-α-carbon thioether crosslinks.
- Sactipeptides are particularly challenging because they contain non-canonical crosslinks that are underrepresented in existing structural datasets, making them an ideal benchmark for testing how well deep learning models generalize beyond evolutionary priors.
- The study introduces a new zero-shot benchmark for evaluating the ability of structure prediction models to correctly predict the 3D conformations of sactipeptides, specifically the geometry of their thioether crosslinks.
- Six leading protein structure predictors—AlphaFold 2, AlphaFold 3, Boltz-1, ESMFold, OmegaFold, and RoseTTAFold 2—were tested on 10 known sactipeptides, five of which have experimentally resolved structures.
- Results show that all models exhibit poor performance in correctly predicting sactibonds, with an average GDT-TS of only 11.5% for known sactipeptides and 12.6% for unknown ones. None of the models achieved crosslink distances near the experimentally observed 1.8 Å sulfur-to-α-carbon bond length.
- Boltz-1 and RoseTTAFold 2 achieved the highest GDT-TS scores, suggesting that some models partially capture local geometric constraints. However, most models incorrectly predicted disulfide bonds instead of the required thioether crosslinks, highlighting their reliance on evolutionary priors.
- Visual analysis reveals that models frequently collapse sulfur atoms into unnatural conformations, demonstrating systematic biases in deep learning-based protein structure predictors when faced with non-standard modifications.
- The findings suggest that future structure prediction methods must integrate physics-informed modeling and explicitly account for rare PTMs to achieve accurate 3D structure predictions beyond standard evolutionary data.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinFolding #StructuralBiology #DeepLearning #Bioinformatics #PostTranslationalModifications #AI
1
14
57
4,230