ViralMap: Predicting Features in Viral Proteins from Primary Sequence
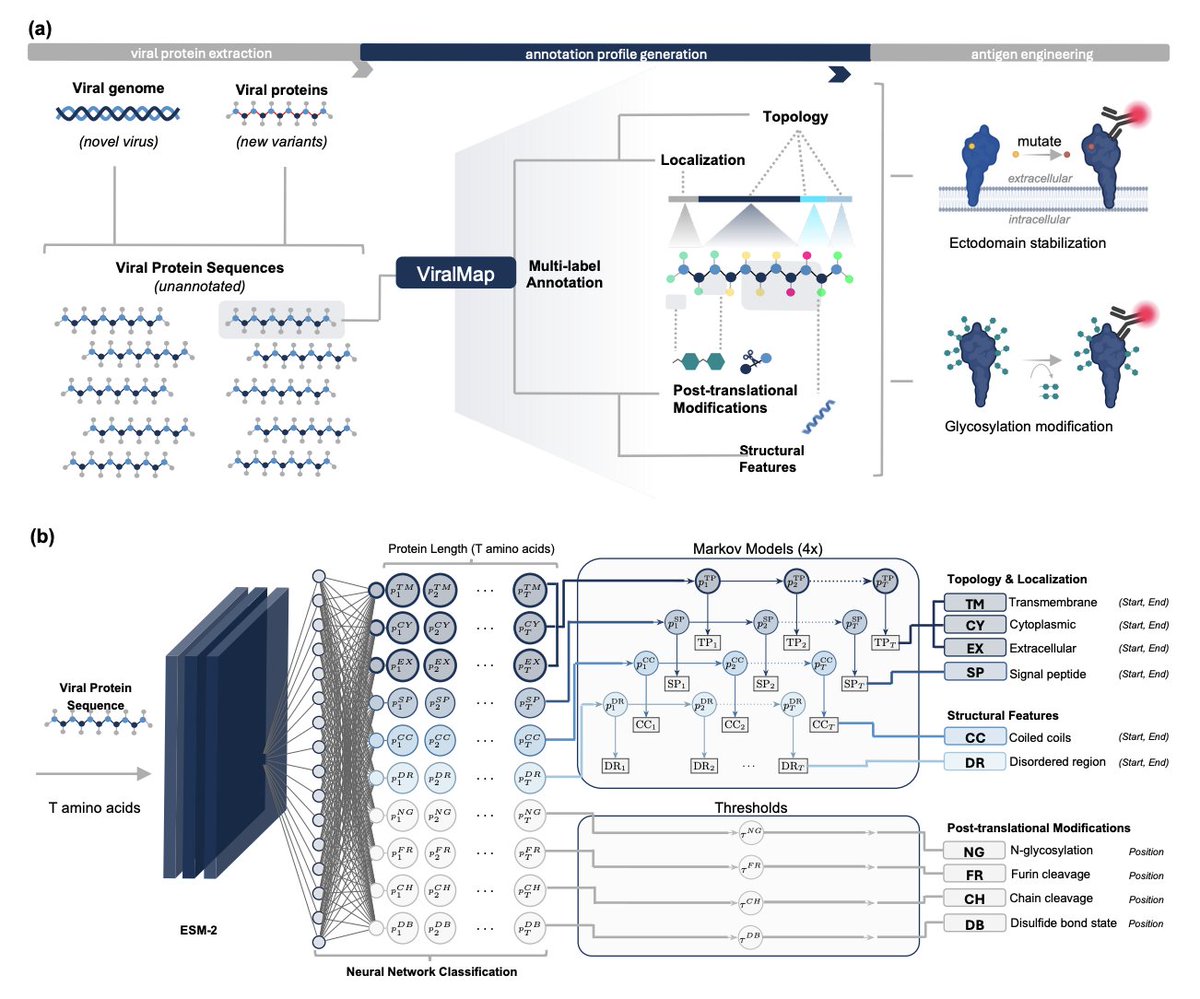
1 ViralMap is presented as a single multi-label model that converts an unannotated eukaryotic viral protein sequence into a residue-level “annotation profile” spanning 10 feature classes relevant to antigen engineering, aiming to replace fragile multi-tool pipelines used in rapid vaccine design workflows.
2 The 10 predicted classes cover three engineering-relevant categories: topology/localization (signal peptide, transmembrane, cytoplasmic, extracellular), post-translational modifications (N-glycosylation, furin cleavage, chain cleavage boundaries, disulfide-bonded cysteines), and structural features (coiled coils, intrinsically disordered regions).
3 Architecture: a two-stage pipeline combining ESM-2 (33-layer, 650M) residue embeddings with a small neural classification head (only last 4 transformer layers unfrozen), followed by structured post-processing—HMM/Viterbi decoding for region-like annotations (topology, signal peptide, coiled coil, disorder) and F2-optimized thresholds for site-like annotations (glycosylation, cleavage, disulfide state).
4 A key contribution is annotation-aware viral dataset curation from UniProt at scale: starting from ~4.1M non-phage viral proteins, the authors filter by annotation quality and length, cluster sequences (MMseqs2), then select cluster representatives using an information-gain strategy that explicitly scores evidence quality (ECO-derived) and seeks complementary annotation coverage across the 10 tasks, yielding 8,238 proteins (~3.13M residues).
5 Evaluation uses 5-fold cross-validation with viral-family-aware splitting plus strict homology separation to probe both cross-strain generalization and novel-family generalization; performance is reported at residue level, with PR-AUC for ViralMap and precision/recall comparisons against established tools where available.
6 ViralMap reports PR-AUC ≥ 0.75 for 7/10 classes, and competitive or better precision/recall than specialized tools across multiple tasks on eukaryotic viral proteins, including: higher precision and recall than NetNGlyc for N-glycosylation, markedly higher precision than ProP for furin cleavage, and much higher precision than AIUPred for disordered regions (with a more balanced precision/recall tradeoff).
7 The results argue ViralMap is not merely motif-scanning: only a minority of canonical motifs are annotated as functional in UniProt (e.g., Asn-X-Ser/Thr for glycosylation; Arg-X-Lys/Arg-Arg for furin), yet performance remains similar when evaluation is restricted to canonical motifs—suggesting contextual sequence information from the language model contributes substantially.
8 Case studies emphasize antigen-design relevance on complex glycoproteins: for SARS-CoV-2 Spike (a Coronaviridae family held out; also longer than the training length cap), ViralMap recovers signal peptide/extracellular/TM/cytoplasmic topology, identifies the S1/S2 furin site and S2’ site, localizes HR1/HR2 coiled-coil fusion machinery, captures all 22 reference N-glycosylation sites, and identifies most disulfide-bonded cysteines (with minor overprediction); it struggles on Spike disordered regions, highlighting limits in novel-family generalization.
9 For HIV-1 Env gp160 (Retroviridae split across folds, testing cross-strain generalization), ViralMap predicts topology and the gp120/gp41 furin cleavage boundary, identifies all 29 reference N-glycosylation sites, recovers most disulfide-bonded cysteines, and localizes a key gp41 coiled-coil region plus an annotated intrinsically disordered segment—features directly tied to fusion and epitope exposure strategies.
💻Code: github.com/HMRI-ADAPT/vmap
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #Bioinformatics #ProteinLanguageModels #Virology #VaccineDesign #AntigenEngineering #ESM2 #SequenceAnnotation #MachineLearning #PandemicPreparedness
4
21
1,684

7 May 2025
MAP Format for Representing Chemical Modifications, Annotations, and Mutations in Protein Sequences: An Extension of the FASTA Format
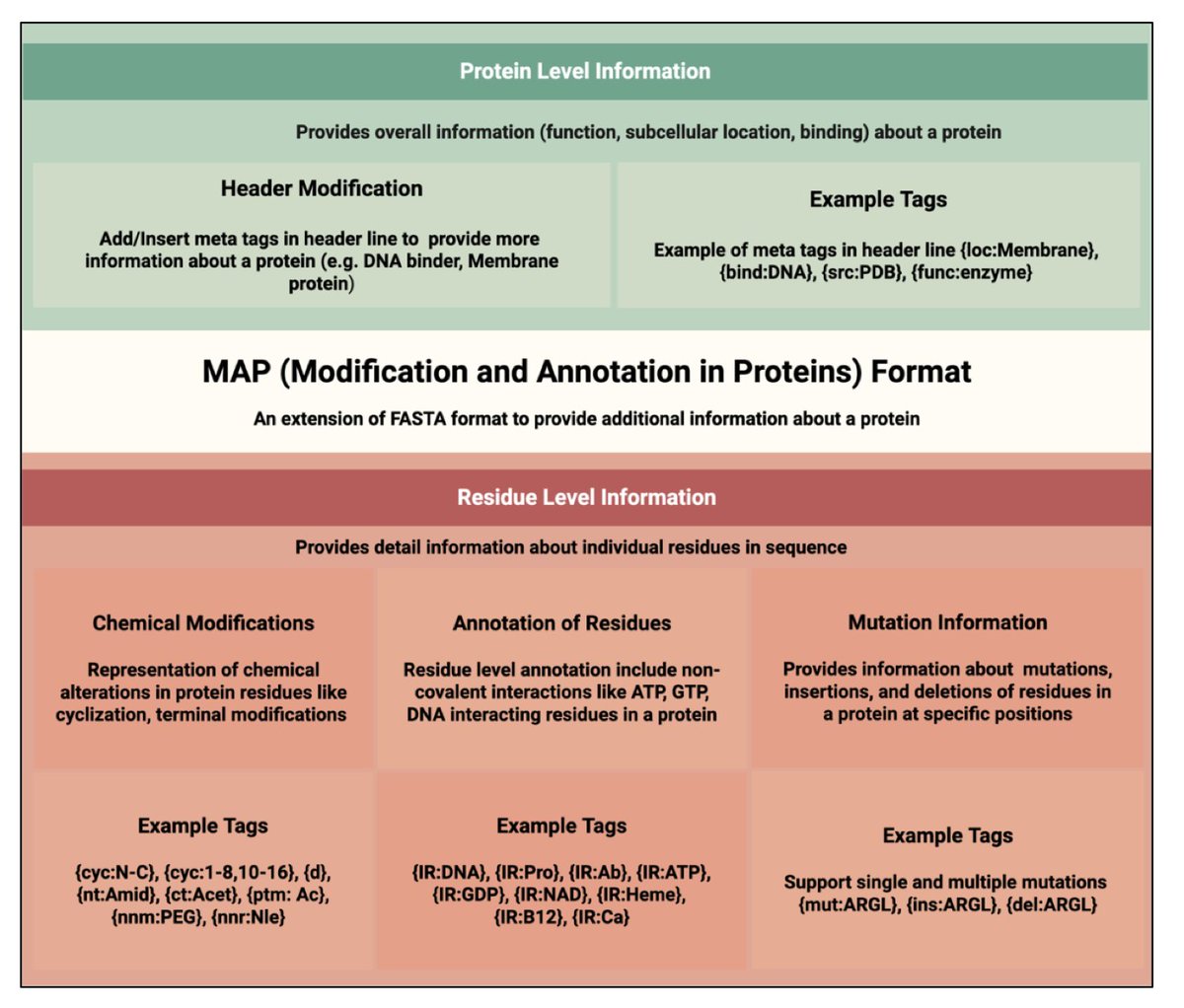
1. This study proposes MAP, a new sequence format that extends the widely used FASTA format to include in-line annotations for protein modifications, mutations, and structural features—retaining readability and compatibility with existing bioinformatics tools.
2. MAP uses intuitive curly-brace tags embedded directly within the protein sequence to denote modifications like phosphorylation, cyclization, non-natural residues, or D-amino acids, providing residue-level annotation in a single-line, human-readable format.
3. Header metadata is structured using tags like {org\:Homo sapiens} or {func\:DNA-binding}, allowing easy parsing of protein-level attributes like organism, subcellular location, and function—enhancing integration with protein databases.
4. Unlike PEFF, HELM, or BILN, which are either too simplistic or overly complex, MAP strikes a balance—simple enough for biologists to edit manually, yet expressive enough to represent therapeutic peptides and engineered proteins with chemical modifications.
5. MAP can denote sequence variants such as mutations (e.g., {mut\:R}), insertions ({ins\:MK}), and deletions ({del\:RTH}) within the linear sequence, enabling unified representation of wild-type and mutant forms for variant annotation and analysis.
6. The format supports tagging of interacting residues using {IR\:Target} (e.g., H{IR\:Zn}), enabling capture of functional sites like metal ion binding or nucleic acid interaction sites directly within the sequence.
7. Applications span protein therapeutics, synthetic biology, structural bioinformatics, and database curation. MAP is already positioned to enhance resources like UniProt, PDB, and peptide therapeutic databases by embedding rich residue-level context.
8. MAP is backward-compatible: removing all curly-brace tags produces a valid FASTA sequence, ensuring that legacy tools can still interpret core sequence content while MAP-aware tools leverage annotations.
9. The authors provide a web portal (MAPrepo) offering manuals, curated datasets, database exports, and Python scripts for converting between formats, facilitating immediate adoption by the community.
10. MAP’s simplicity, flexibility, and compatibility position it as a practical standard for next-generation protein sequence annotation—bridging raw sequences and chemically informed representations in a unified, FAIR-compliant format.
💻Code: webs.iiitd.edu.in/raghava/ma…
📜Paper: arxiv.org/abs/2505.03403
#ProteinSequence #FASTA #MAPformat #Bioinformatics #ProteinModification #PostTranslationalModifications #DataStandards #Proteomics #ComputationalBiology #SequenceAnnotation #TherapeuticProteins #FAIRdata

1
2
670
7 May 2025
MAP Format for Representing Chemical Modifications, Annotations, and Mutations in Protein Sequences: An Extension of the FASTA Format
1. This study proposes MAP, a new sequence format that extends the widely used FASTA format to include in-line annotations for protein modifications, mutations, and structural features—retaining readability and compatibility with existing bioinformatics tools.
2. MAP uses intuitive curly-brace tags embedded directly within the protein sequence to denote modifications like phosphorylation, cyclization, non-natural residues, or D-amino acids, providing residue-level annotation in a single-line, human-readable format.
3. Header metadata is structured using tags like {org\:Homo sapiens} or {func\:DNA-binding}, allowing easy parsing of protein-level attributes like organism, subcellular location, and function—enhancing integration with protein databases.
4. Unlike PEFF, HELM, or BILN, which are either too simplistic or overly complex, MAP strikes a balance—simple enough for biologists to edit manually, yet expressive enough to represent therapeutic peptides and engineered proteins with chemical modifications.
5. MAP can denote sequence variants such as mutations (e.g., {mut\:R}), insertions ({ins\:MK}), and deletions ({del\:RTH}) within the linear sequence, enabling unified representation of wild-type and mutant forms for variant annotation and analysis.
6. The format supports tagging of interacting residues using {IR\:Target} (e.g., H{IR\:Zn}), enabling capture of functional sites like metal ion binding or nucleic acid interaction sites directly within the sequence.
7. Applications span protein therapeutics, synthetic biology, structural bioinformatics, and database curation. MAP is already positioned to enhance resources like UniProt, PDB, and peptide therapeutic databases by embedding rich residue-level context.
8. MAP is backward-compatible: removing all curly-brace tags produces a valid FASTA sequence, ensuring that legacy tools can still interpret core sequence content while MAP-aware tools leverage annotations.
9. The authors provide a web portal (MAPrepo) offering manuals, curated datasets, database exports, and Python scripts for converting between formats, facilitating immediate adoption by the community.
10. MAP’s simplicity, flexibility, and compatibility position it as a practical standard for next-generation protein sequence annotation—bridging raw sequences and chemically informed representations in a unified, FAIR-compliant format.
💻Code: webs.iiitd.edu.in/raghava/ma…
📜Paper: arxiv.org/abs/2505.03403
#ProteinSequence #FASTA #MAPformat #Bioinformatics #ProteinModification #PostTranslationalModifications #DataStandards #Proteomics #ComputationalBiology #SequenceAnnotation #TherapeuticProteins #FAIRdata
2
549