
11 Jul 2025
Microsoft’s new AI system to help decode protein motion, advance drug discovery: Satya Nadella yespunjab.com/?p=138801
#MicrosoftAI #BioEmu #DrugDiscovery #AIinScience #ProteinMotion #SatyaNadella #DeepLearning #AIResearch #BiomolecularEmulator #ProteinStructures #AIDrugDevelopment
@Microsoft
46

17 Jun 2025
✨Day 2 of the #AlphaFoldWorkshopUG2025 at @IDIMakerere, @Makerere!
Today's sessions covered:
🔹 Introduction to #AlphaFold
🔹 Assessing the quality of predicted #proteinstructures
🔹 Accessing & engaging with #AlphaFold
This workshop is supported by @GoogleDeepMind.
#AlphaFold




14
30
1,229

4 Mar 2025
🧬 Exciting AlphaFold DB updates:
✅ Bulk downloads – Up to 100 structures (PDB, mmCIF / JSON)
✅ pLDDT filtering – Find high-confidence models
✅ Table exports
📖 More
sciencedirect.com/science/ar…
🔗 Explore
alphafold.ebi.ac.uk
#AlphaFold #Bioinformatics #ProteinStructures

1
19
53
2,840
3 Mar 2025
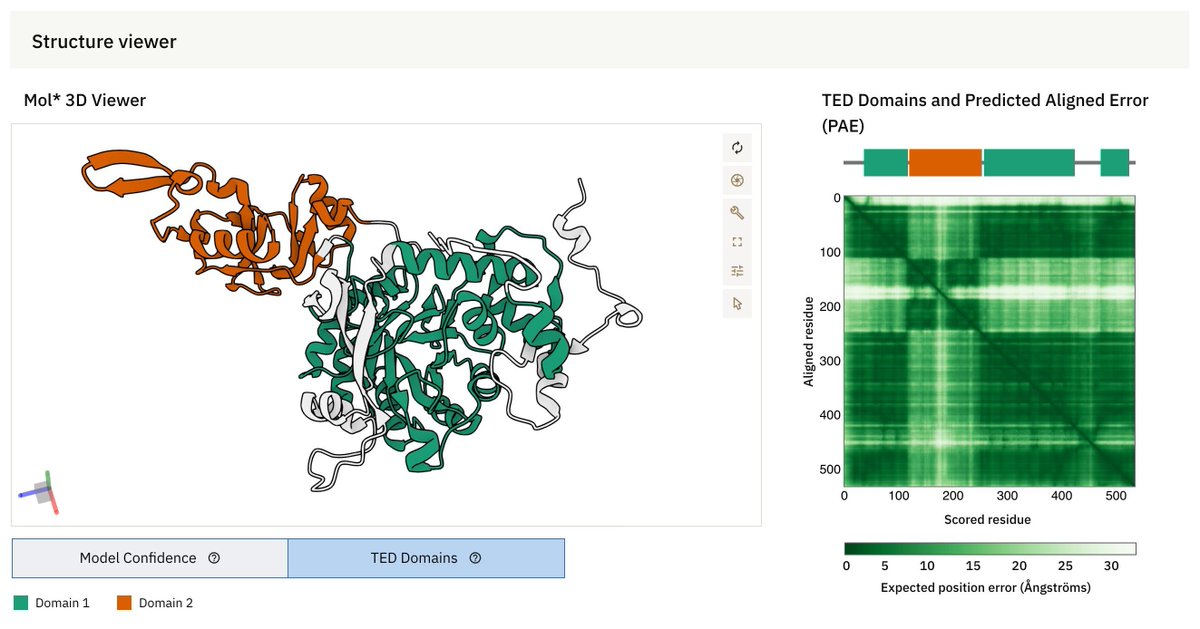
🚀 New in AlphaFold DB: Structural domains at a glance! 🔍✨
AFDB now integrates TED (The Encyclopedia of Domains) to classify functional protein domains.
🔗 More
ebi.ac.uk/about/news/updates…
🔗 Explore
alphafold.ebi.ac.uk
#AlphaFold #TED #ProteinStructures
4
57
181
13,015

15 Jan 2025
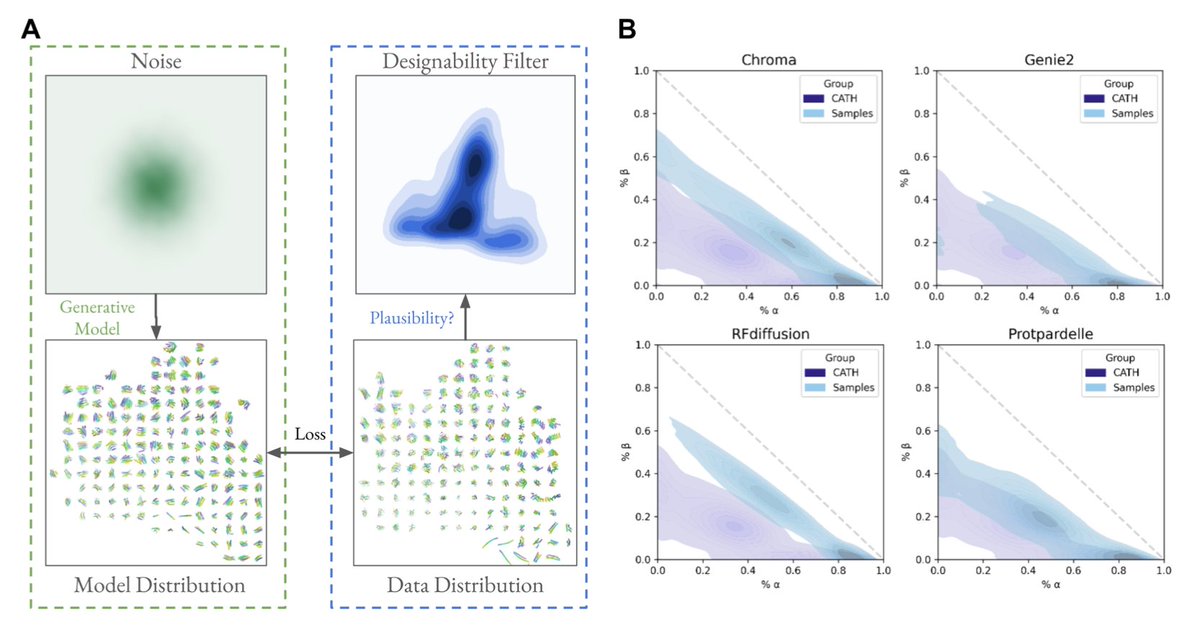
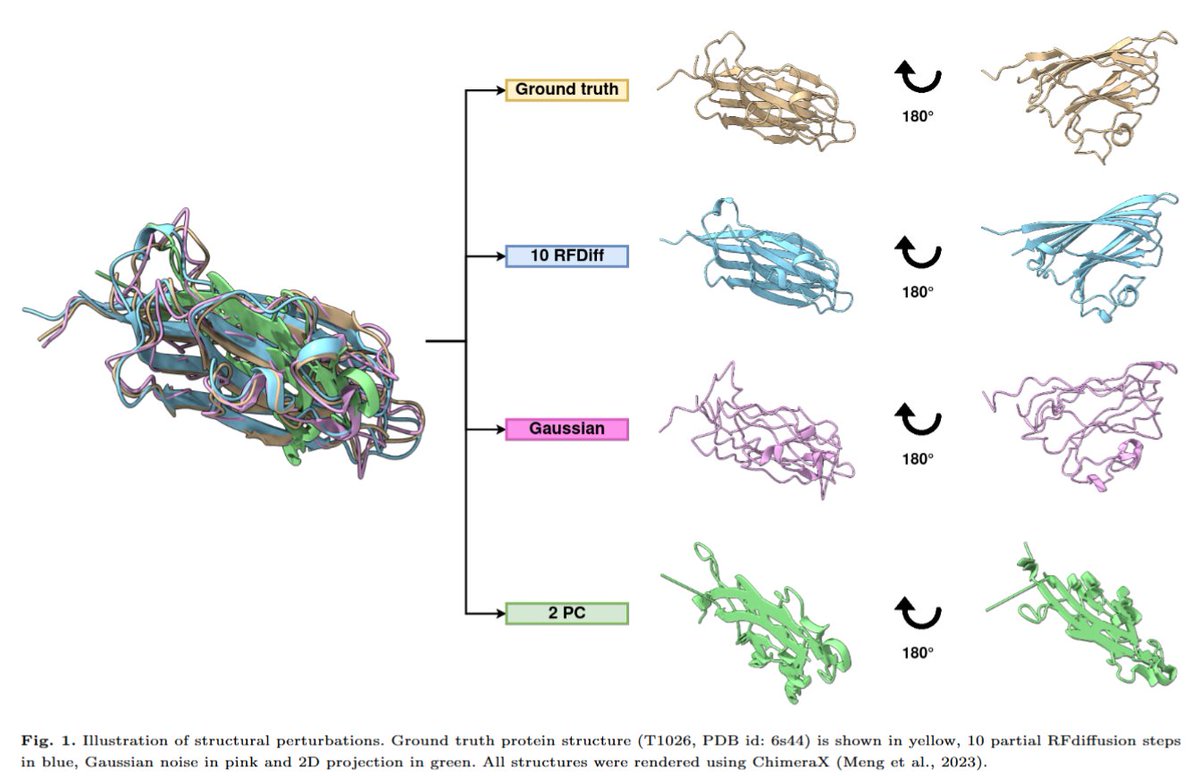
Assessing Generative Model Coverage of Protein Structures with SHAPES
1. This paper introduces SHAPES, a comprehensive framework to evaluate generative protein structure models by assessing their coverage of the protein structure space, highlighting regions often undersampled or biased.
2. SHAPES uses structural embeddings and Fréchet Protein Distance (FPD) to quantitatively compare generated protein structures with experimentally validated databases like CATH, revealing systematic biases in generative model outputs.
3. A key finding is that current generative models favor highly designable structures, often over-idealized with excessive alpha helices and beta sheets, while undersampling flexible motifs like loops and mixed alpha-beta domains critical for functional diversity.
4. The study benchmarks five generative models, including Chroma, RFdiffusion, Genie2, Multiflow, and Protpardelle, showing distinct biases and capabilities in structural coverage depending on sampling noise scales and model architecture.
5. Increased sampling temperature and noise scales improve structural diversity but at the expense of designability, exposing a trade-off between creating diverse structures and maintaining practical design utility.
6. The researchers identify functional tertiary motifs (TERMs) absent in most generative model outputs, emphasizing the need for more inclusive models to capture rare yet biologically significant structures.
7. SHAPES establishes itself as a superior metric compared to traditional diversity measures, providing fine-grained insights into where generative models succeed or fail in replicating the natural protein structure universe.
8. The paper advocates for integrating improved sequence design and structure prediction methods to guide models toward more balanced and functional structural outputs.
@PossuHuangLab @zhkim216 @Tianyu_Lu
💻Code: github.com/ProteinDesignLab/…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinDesign #GenerativeModels #MachineLearning #Bioinformatics #ProteinStructures #SHAPESFramework
1
7
33
2,983
21 Dec 2024
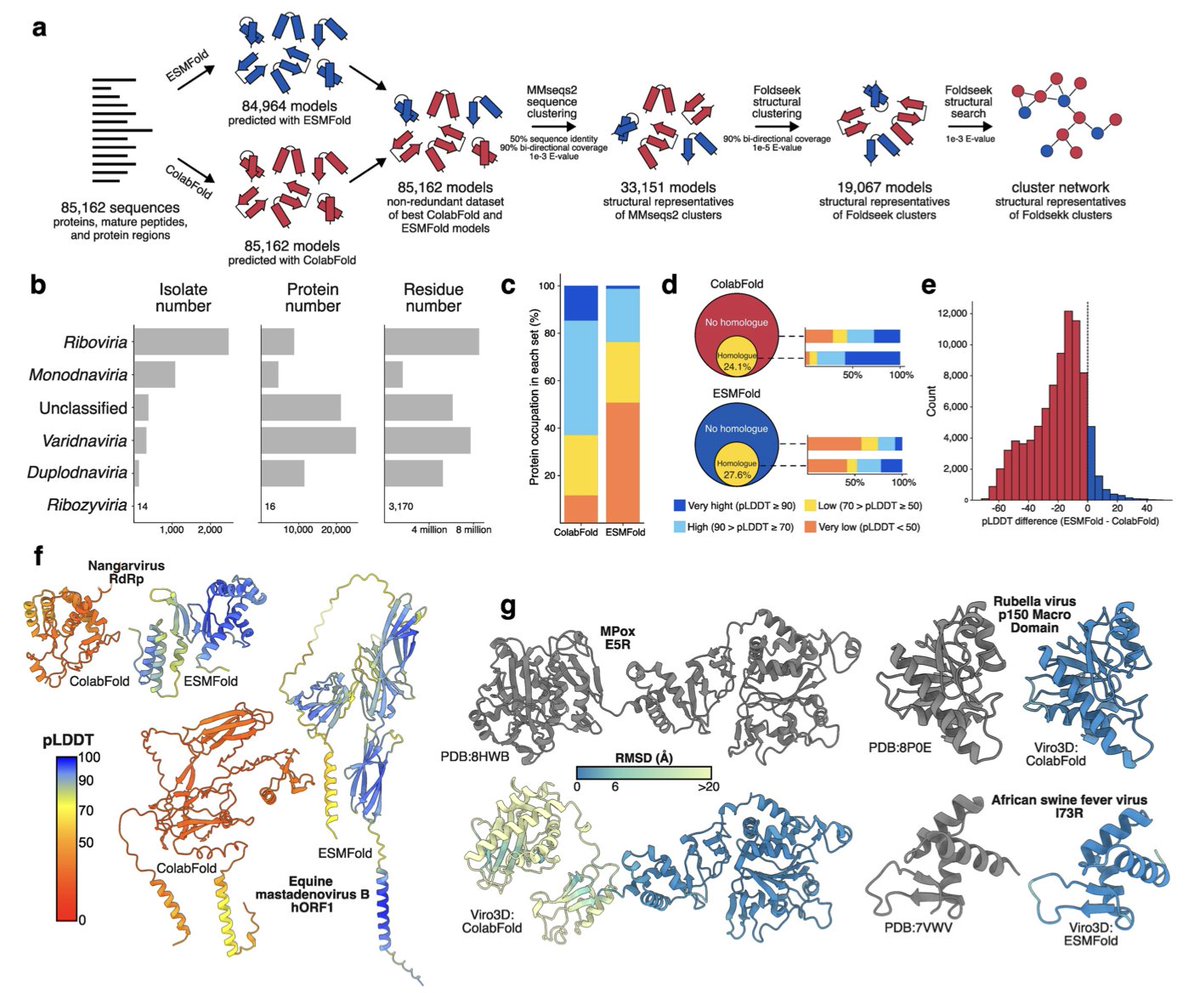
Viro3D: a comprehensive database of virus protein structure predictions
1. The Viro3D database represents a groundbreaking expansion in viral protein structural data, predicting structures for 85,000 proteins across 4,400 viruses. This is a 30-fold increase compared to experimental structures.
2. Leveraging state-of-the-art prediction tools, ColabFold and ESMFold, Viro3D achieved structural predictions with confidence scores above 70 for 64% of the proteins, providing unprecedented coverage and quality.
3. ColabFold outperformed ESMFold in overall prediction accuracy but ESMFold demonstrated advantages for certain long proteins and cases with limited homologous sequences.
4. Beyond predictions, Viro3D uses clustering and network analysis to group proteins based on structural similarity, enabling insights into evolutionary relationships and functional annotation.
5. Highlighting structural conservation, Viro3D identifies hallmark viral proteins, such as RNA-dependent RNA polymerases and fusion glycoproteins, across diverse viral families.
6. The database sheds light on the deep evolutionary history of viral proteins, including tracing the origins of the coronavirus spike glycoprotein to ancient genetic exchanges with herpesviruses.
7. Viro3D facilitates studies in molecular virology, virus evolution, and therapeutic design, offering an intuitive interface for browsing, visualizing, and downloading protein models.
@GroveLab @SpyrosLytras @UladLitvin
💻Code: viro3d.cvr.gla.ac.uk
📜Paper: biorxiv.org/content/10.1101/…
#Virology #StructuralBiology #ProteinStructures #Bioinformatics #VirusEvolution
21
87
6,703
21 Dec 2024
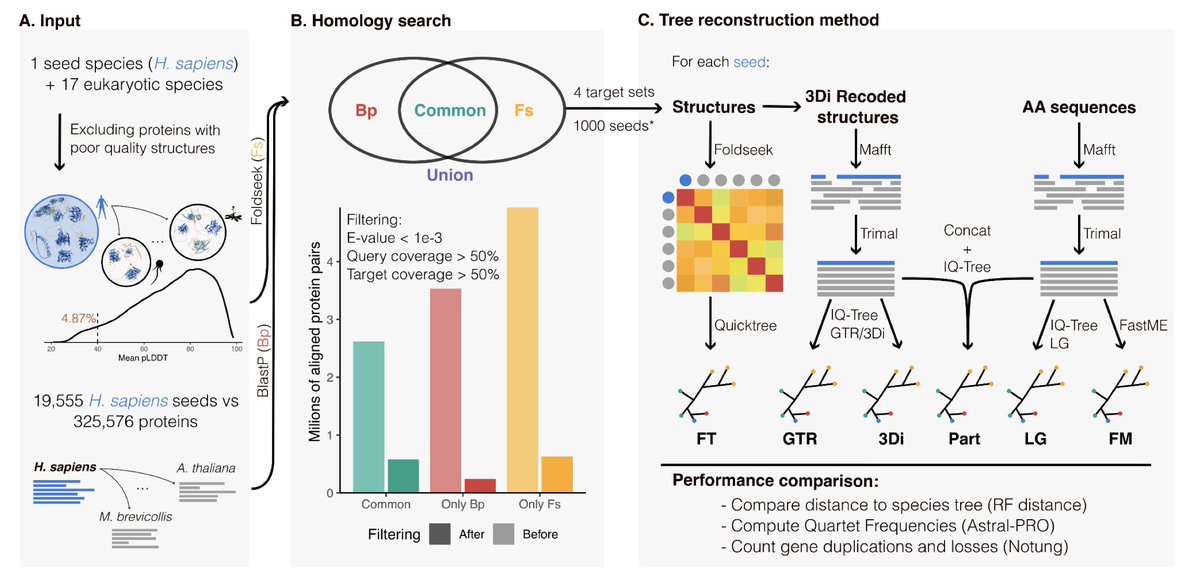
Newly developed structure-based methods do not outperform standard sequence-based methods for large-scale phylogenomics
1. The study evaluates whether newly developed structure-based methods for phylogenetics, such as Foldseek and Foldtree, surpass traditional sequence-based methods. Surprisingly, structure-based approaches fail to outperform sequence-based methods in reconstructing large-scale phylogenies.
2. Structure-based tools like Foldseek excel at detecting remote homologs in the "twilight zone" of sequence identity, but they miss many homologs identified by sequence-based tools like BlastP, leading to higher false positive rates.
3. Sequence-based maximum likelihood (ML) methods such as LG consistently outperform all structure-based methods in tree accuracy metrics, including lower Robinson-Foulds distances and higher quartet support.
4. Structure-based methods show promise in certain niche applications, leveraging AlphaFold-generated protein models, but their computational demands and limited phylogenetic accuracy suggest significant room for improvement.
5. A hybrid approach combining sequence and structural data did not outperform sequence-based ML methods alone, underscoring the current limitations of structural methods in large-scale studies.
6. The study highlights the importance of continuing to refine structural data encoding and computational pipelines to potentially unlock the full potential of structure-based phylogenetics in the future.
@toni_gabaldon @giacomutti
💻Code: github.com/Gabaldonlab/struc…
📜Paper: biorxiv.org/content/10.1101/…
#Phylogenomics #ComputationalBiology #AlphaFold #ProteinStructures #Bioinformatics
5
16
2,048

19 Dec 2024
🔬How does #AlphaFold2 perform with minimal sequence input?
"Dissecting AlphaFold2’s capabilities with limited sequence information" reveals insights into its ability to predict #ProteinStructures using structural templates without deep MSAs.
Read more: doi.org/10.1093/bioadv/vbae1…
2
2
398
17 Dec 2024
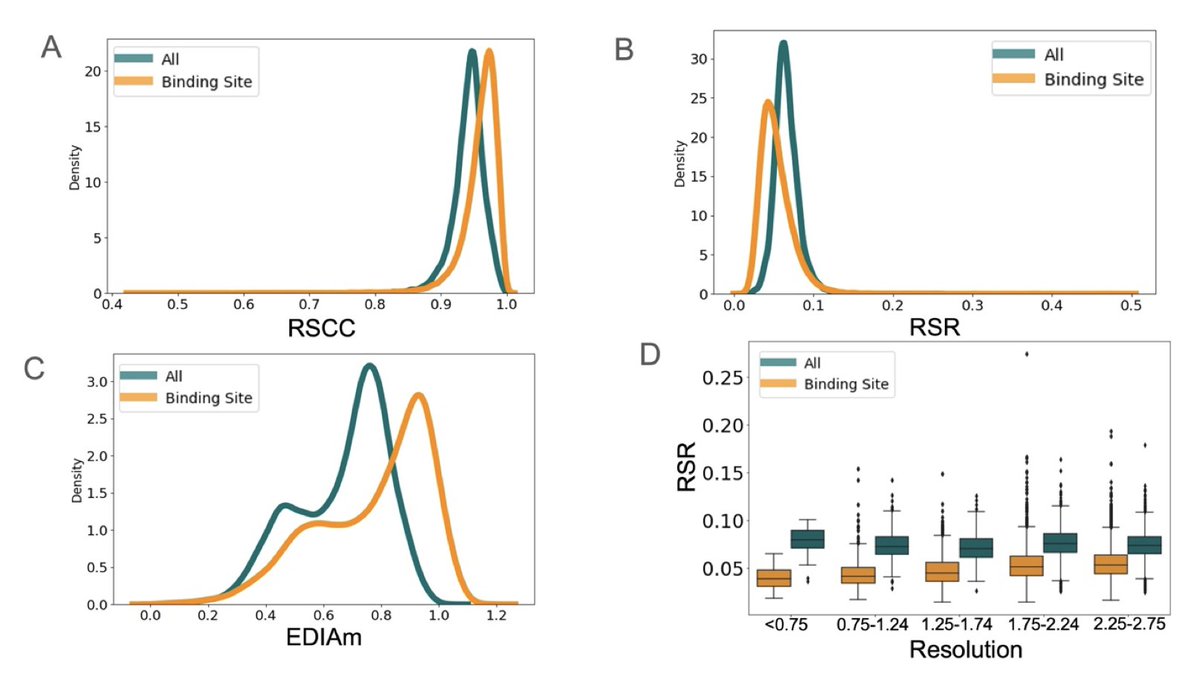
Modeling Bias Toward Binding Sites in PDB Structural Models
1. Protein structural models from the PDB, central to biology and machine learning, show significant modeling biases: binding sites are better modeled and fit the experimental data more accurately than non-binding regions.
2. Using metrics like RSCC, RSR, and EDIAm to measure data fit, binding site residues consistently perform better, with higher RSCC (0.96 vs. 0.94) and lower RSR (0.058 vs. 0.076), revealing a focus on "important" regions during manual modeling.
3. These trends persist regardless of resolution or Rfree values, showing that global model quality metrics fail to eliminate local biases in how structures are refined.
4. Binding site residues are more likely to have alternative conformations (5.0% vs. 1.9% elsewhere), indicating that modelers pay greater attention to these areas, manually improving their fit to experimental data.
5. Non-ideal side-chain rotamers at binding sites are better supported by electron density, confirming that unusual conformations in binding regions are biologically meaningful and not artifacts of poor modeling.
6. Pocket residues identified in structures without ligands exhibit similar, though less pronounced, trends, suggesting these biases stem from modeling decisions rather than biological differences alone.
7. This bias toward binding sites has profound implications: structural models are used as "truth" in simulations, docking studies, and machine learning algorithms. Overlooking non-binding regions may propagate errors in downstream analyses.
8. Recognizing these biases highlights the need for improved automated modeling techniques and local metrics to validate entire protein structures, ensuring unbiased biological interpretations and reliable machine learning inputs.
@stephanie_mul
📜Paper: biorxiv.org/content/10.1101/…
#ProteinStructures #PDB #StructuralBiology #MachineLearning #BiasInData #ComputationalBiology
1
1
12
1,852
17 Nov 2024
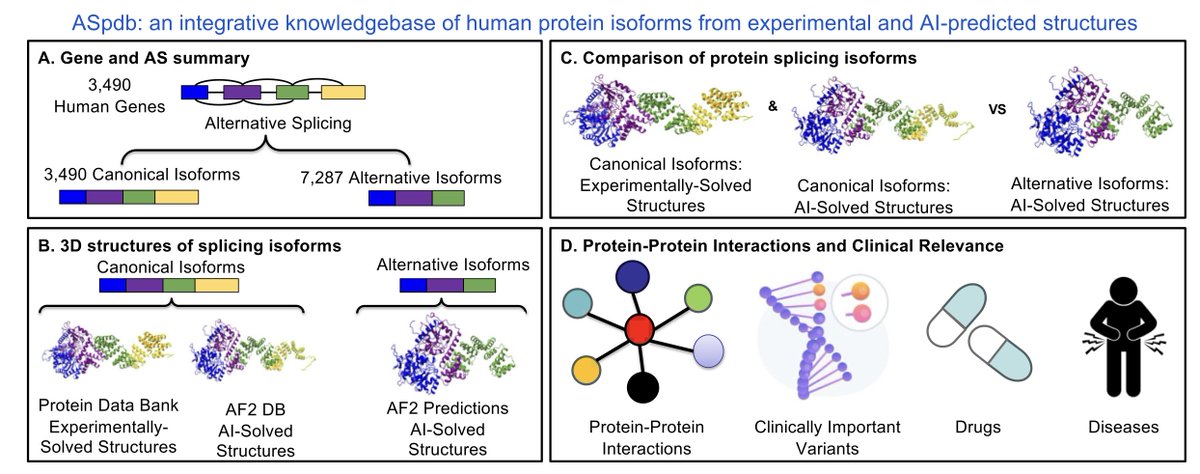
ASpdb: An Integrative Knowledgebase of Human Protein Isoforms from Experimental and AI-Predicted Structures
1. ASpdb is a groundbreaking database combining experimental and AlphaFold2-predicted protein structures for over 10,000 human protein isoforms, including both canonical and alternative splicing isoforms.
2. The database uniquely integrates 3D structural data with sequence variations, splicing events, and functional annotations, enabling comparative analyses of structural alterations caused by alternative splicing.
3. Over 7,200 alternative isoforms are included, featuring detailed evaluations of AlphaFold2 predictions using multiple reliability metrics like pLDDT scores and TM-scores.
4. ASpdb links structural variations to clinical insights by incorporating data on protein-protein interactions, disease associations, and drug interactions, offering a multidimensional resource for translational research.
5. Key innovations include statistical tests for structural differences between isoforms, visualization tools for splicing-induced changes, and integration of gene expression profiles from TCGA and GTEx databases.
6. The database addresses gaps left by other resources, such as lack of structural data in cancer-specific splicing databases (OncoSplicing, ASCancer Atlas) and limited analysis capabilities in structural repositories (CHESS, APPRIS).
7. ASpdb is optimized for studying the impact of alternative splicing on diseases like cancer and Alzheimer’s, facilitating the identification of isoform-specific biomarkers and therapeutic targets.
@PoraKim99 @HongyuZhao2 @AlbonWu
📜Paper: academic.oup.com/nar/advance…
#AlternativeSplicing #Proteomics #AlphaFold2 #Bioinformatics #ProteinStructures #ClinicalResearch
6
24
1,970
16 Nov 2024
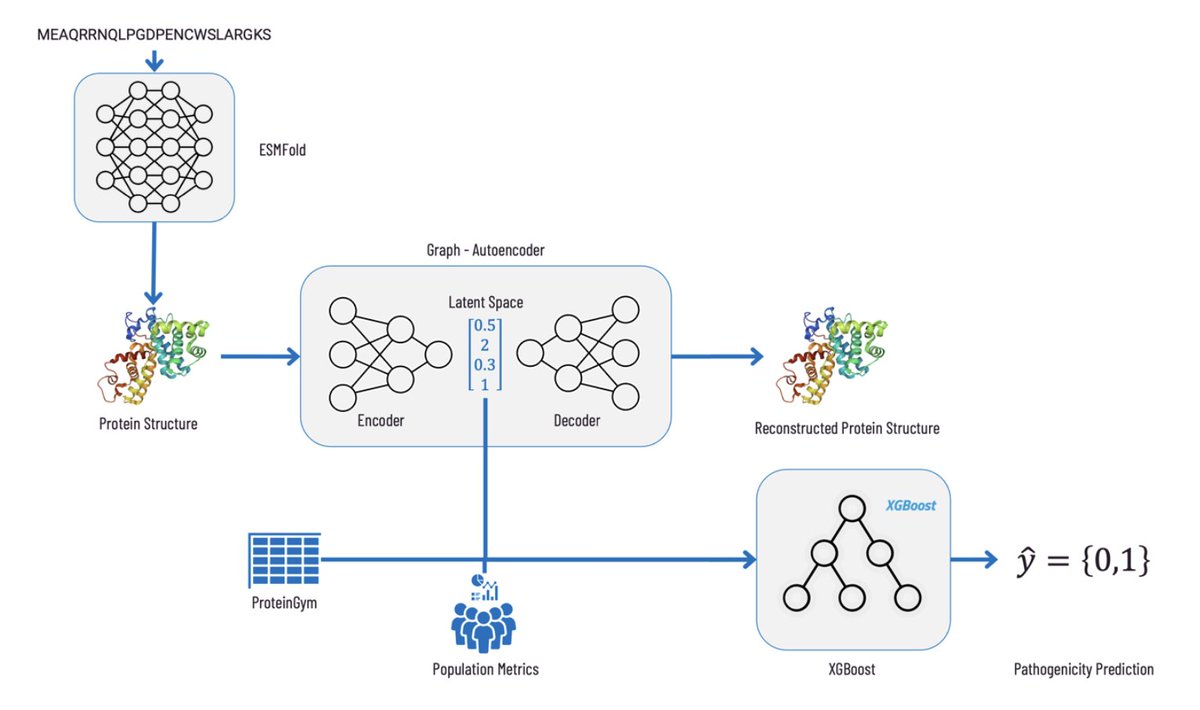
Utilizing protein structure graph embeddings to predict the pathogenicity of missense variants
• The study introduces a machine learning workflow leveraging graph embeddings derived from protein structures predicted by ESMFold to classify the pathogenicity of missense variants.
• Structural embeddings are generated using graph autoencoders at two levels: residue and atomic. These embeddings capture both local and global structural information for enhanced pathogenicity prediction.
• A combined model utilizing both residue- and atomic-level embeddings achieves the highest accuracy, with an AUROC of 0.924, outperforming models trained without structural data.
• The workflow integrates graph embeddings with population metrics from gnomAD and functional data from ProteinGym, offering a holistic approach to variant classification.
• Structural information significantly boosts the performance of XGBoost classifiers, demonstrating the utility of protein structure data in pathogenicity predictions.
• Embedding size experiments reveal that smaller embeddings (128 dimensions) consistently outperform larger ones (256 dimensions), ensuring efficient model performance.
• SHAP analysis highlights allele frequency as the most critical feature, followed by wild-type and variant structural embeddings, underscoring the importance of integrating structural insights.
• This method advances the field by replacing manual feature engineering with automated graph-based embeddings, enabling scalable and interpretable predictions for genetic variant analysis.
📜Paper: biorxiv.org/content/10.1101/…
#MissenseVariants #ProteinStructures #Bioinformatics #MachineLearning #PathogenicityPrediction
1
2
11
1,794

VUStruct is a powerful tool that analyzes genetic variants in 3D protein structures, revolutionizing rare disease diagnostics and research! #RareDiseases #ProteinStructures
📄 doi.org/10.1101/2024.08.06.6…
EVBC👤: @MeilerLab
4
358

19 Sep 2024
Genomics 2 Proteins portal: a resource and discovery tool for linking genetic screening outputs to protein sequences and structures. #GenericVariants #ProteinStructures @naturemethods nature.com/articles/s41592-0…
7
11
764

10 Sep 2024
Our study with @rogoulenko in @NAR_Open shows #DNA flexibility impacts protein binding. Despite kinetic challenges, evolution ensures proteins effectively target and bind DNA. Check out Daniel Philosoph's cover art, hand-painted with real brushes! #ProteinDNA #ProteinStructures

3
3
363

3 Jul 2024
Happy to start working with new 1,2 GHz NMR spectrometer - perfect for precise analysis of complex biological systems like #proteinstructures ! Peter Schmieder, @HanSun_lab , Adam Lange, @MillesSigrid , Hartmut Oschkinat
Read more 👇
leibniz-fmp.de/newsroom/news…

12
30
2,095
31 Mar 2024
Happy Easter to all who are celebrating!
A chick made from lysozyme, a very abundant protein in the PDB (1000 structures).
#Easter #ProteinStructures #PDB #PDBe
buff.ly/3VBByID

1
3
33
3,328

29 Jan 2024
🎉Exciting news! Our latest paper in @NatureComms delves into protein-protein interaction networks & IDRs using AlphaFold2. Dive into our findings here! 🧬#Bioinformatics #ProteinStructures
1/8
ALT Robot hand placing a protein fragment into the surface of a globular protein.
5
44
253
35,387
29 Nov 2023
Using machine learning to predict the structure of proteins that bind to DNA and RNA. #MachineLearning #ProteinDNA #ProteinStructures @naturemethods
nature.com/articles/s41592-0…
29
109
9,186

28 Nov 2023
💭 What can NMR Spectroscopy reveal about protein structures?
buff.ly/3GkvpIc
#ProteinStructures #StructuralBiology #NMRSpectroscopy #Proteins #ProteinAnalysis
2
3
469

7 Nov 2023
Calling all Oncode researchers! Dive into the fascinating world of #ProteinStructures and molecular interactions at our upcoming Masterclass on December 8th! Organized by @TassosPerrakis & @TitiaSixma. More info & registration via oncodeinstitute.nl/events/ma…
3
1,465