
23 Jun 2025
Consistent Sampling and Simulation: Molecular Dynamics with Energy-Based Diffusion Models
1.Recent work highlights a core challenge in molecular simulation with diffusion models: while these models can sample equilibrium states well, they fail to produce consistent results when used for molecular dynamics (MD) simulations. This inconsistency stems from a violation of the Fokker-Planck equation at small diffusion times.
2.To address this, the authors introduce a Fokker-Planck-based regularization that enforces consistency between sampling and simulation by aligning the model’s learned score (energy gradient) with the Fokker-Planck equation.
3.They design an energy-based diffusion model that is conservative (score = energy gradient), enabling stable and physically meaningful MD simulations without requiring force labels—just equilibrium samples.
4.A key innovation is the use of a computationally efficient "weak" residual formulation of the Fokker-Planck equation. This avoids costly high-order derivatives and enables scalable training using only first-order computations.
5.To further improve training efficiency, they introduce a Mixture of Experts (MoE) framework that partitions the diffusion time into intervals. A separate model is trained for each interval, applying the Fokker-Planck regularization only where needed (small t), which enhances performance and reduces compute.
6.They demonstrate their method across a range of settings: a toy Müller-Brown potential, alanine dipeptide, and a large-scale transferable model across 400 dipeptides. In all cases, their method achieves superior consistency between sampling and simulation.
7.For alanine dipeptide, their model (Both: MoE Fokker-Planck regularization) recovers important conformational states missed by standard diffusion models, while preserving structural fidelity, unlike competing methods that inject simulation noise.
8.On the full dipeptide benchmark, their approach achieves state-of-the-art results, significantly improving the agreement between Langevin simulations and independent samples, with clear gains in free energy accuracy (measured via JS divergence and PMF error).
9.An ablation analysis reveals that MoE and Fokker-Planck regularization improve consistency via distinct mechanisms, and their combination outperforms either alone—supporting their complementary roles.
10.This work enables energy-based diffusion models to support both accurate equilibrium sampling and consistent MD simulations, paving the way for unified generative and dynamical modeling in coarse-grained biophysics.
💻Code: github.com/noegroup/ScoreMD
📜Paper: arxiv.org/abs/2506.17139v1
#DiffusionModels #MolecularDynamics #ScoreBasedModels #FokkerPlanck #CoarseGraining #ComputationalBiophysics #GenerativeModels

3
713
1 May 2025
ProT-GFDM: A Generative Fractional Diffusion Model for Protein Generation
1. ProT-GFDM introduces a novel generative protein design framework that leverages fractional Brownian motion (fBm) in diffusion models to improve the generation of protein backbone structures by capturing long-range dependencies in data.
2. Unlike traditional generative models relying on standard Brownian motion (BM), ProT-GFDM employs a Markov approximation of fBm (MA-fBm), enabling the modeling of temporal memory effects and non-local correlations, which are essential for protein structural coherence.
3. The model is formulated as a continuous-time score-based diffusion process and can be solved using either stochastic differential equations (SDEs) or their deterministic counterparts, probability-flow ODEs (PF-ODEs), providing flexibility in sampling and inference.
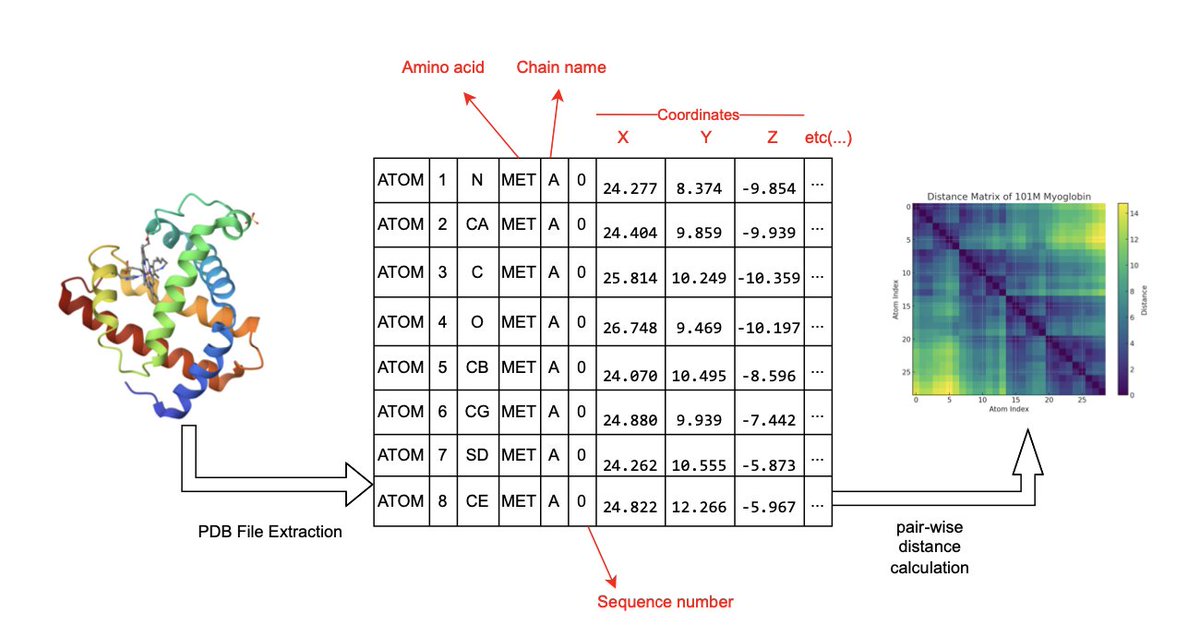
4. ProT-GFDM models protein structures via α-carbon distance maps derived from the Protein Data Bank (PDB), using them as the target representation in a diffusion-based generative pipeline.
5. Experimental evaluations show that ProT-GFDM outperforms classical score-based models (e.g., VP-SDE) with a 7.19% increase in density, 5.66% improvement in coverage, and 1.01% reduction in FID, particularly when using higher Hurst indices and appropriate solvers.
6. The model supports both linear and cosine noise schedules, with the cosine schedule offering better sample fidelity under low Hurst settings and the linear schedule excelling in diversity for high Hurst values (e.g., H=0.8).
7. ProT-GFDM is implemented using a conditional U-Net architecture for score function estimation and employs advanced score-matching techniques—including augmented and sliced score matching—to train with noisy data.
8. A variety of sampling strategies, including Euler–Maruyama, Langevin-based predictor-corrector (PC) samplers, and classical ODE solvers like RK4, are benchmarked to assess performance tradeoffs in speed, fidelity, and diversity.
9. The use of fractional noise processes enables the generation of protein structures that are both more diverse and structurally coherent, pushing the boundaries of deep generative modeling in structural bioinformatics.
10. ProT-GFDM presents a promising step forward in generative protein modeling, combining theoretical rigor in stochastic dynamics with empirical improvements in sample quality and efficiency for applications in protein design and computational drug discovery.
📜Paper: arxiv.org/abs/2504.21092
#ProteinDesign #GenerativeModels #DiffusionModels #FractionalDynamics #Bioinformatics #DeepLearning #ComputationalBiology #ScoreBasedModels #StochasticProcesses #AI4Science
8
738

1 Jun 2021
Gotta Go Fast When Generating Data with Score-Based Models
Jolicoeur-Martineau et al.: arxiv.org/abs/2105.14080
#ArtificialIntelligence #DeepLearning #ScoreBasedModels

11
16