
May 27
𝐉𝐨𝐡𝐧 𝐏𝐞𝐭𝐫𝐨𝐥𝐥𝐢𝐧𝐨 𝐂𝐡𝐞𝐜𝐤𝐬 𝐎𝐮𝐭 𝐭𝐡𝐞 𝐍𝐄𝐗𝐓𝐎𝐑𝐂𝐇 𝐍𝐞𝐱𝐃𝐨𝐭 𝐖𝐋𝟐𝟓—𝐁𝐞𝐜𝐚𝐮𝐬𝐞 𝐒𝐨𝐦𝐞𝐭𝐢𝐦𝐞𝐬 𝐎𝐧𝐞 𝐁𝐞𝐚𝐦 𝐈𝐬𝐧’𝐭 𝐄𝐧𝐨𝐮𝐠𝐡
A compact setup with a big job to do.
In this review, John from The Square Reviews takes a hard look at the 𝗡𝗘𝗫𝗧𝗢𝗥𝗖𝗛 𝗡𝗲𝘅𝗗𝗼𝘁 𝗪𝗟𝟮𝟱, a weapon-mounted light & laser combo built for defensive applications where fast target identification and rapid alignment matter.
The WL25 packs both illumination and aiming capability into a compact footprint designed to stay out of the way—while still being ready when things matter most.
💡 High-output tactical light
🔴 Integrated aiming laser
⚡ Quick-access activation controls
🎯 Compact rail-mounted design
🔋 Rechargeable performance built for defensive readiness
But adding a laser changes the conversation.
Is it genuinely useful? A gimmick? Or something that actually enhances speed and confidence under stress?
John breaks down the controls, ergonomics, visibility, and real-world practicality with the brutally honest approach The Square Reviews is known for.
👉 Watch the full review and decide if the WL25 is the right combo for your setup—or just another flashy attachment fighting for rail space.
#NEXTORCH #NexDotWL25 #WeaponLight #LaserSight #LightAndLaser #TacticalLight #EDCgear #HomeDefense #DefensiveSetup #TacticalGear #FlashlightReview #GearReview #EDClife #PreparednessGear #TheSquareReviews #PetrollinoApproved #NoFluffReview #HonestGearReview #TargetIdentification #DutyGear #RechargeableLight #CCWgear #WESHOOT #WESHOOTUSA #2A #Pro2A #TacticalEDC #EverydayCarry
2
131
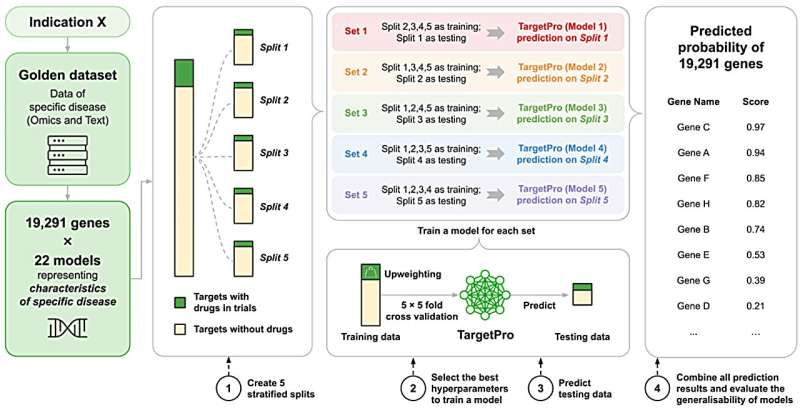
Insilico Medicine, a clinical-stage biotechnology company powered by generative artificial intelligence (AI), recently announced advancements to its unified AI framework for drug target discovery, integrating its previously introduced Target Identification Pro (#TargetPro) and #TargetIdentification Benchmark (TargetBench 1.0) into a validated system designed to improve the accuracy, reliability, and scalability of early-stage drug development.
Published in Scientific Reports, the framework demonstrates strong performance across benchmarking and real-world discovery workflows, reinforcing its role as a standardized foundation for AI-driven target identification.
#Drugdiscovery has long been constrained by high failure rates, with nearly 90% of clinical candidates failing—often due to weak or poorly validated biological targets. Insilico's integrated TargetPro–TargetBench framework addresses this challenge by combining disease-specific predictive modeling with rigorous, standardized evaluation.
Read the latest release here: phys.org/news/2026-04-ai-dru…
2
7
28
6,789

Apr 29
Nature Reviews Drug Discovery | #TargetIdentification and Assessment in the Era of AI
Insilico recently published a comprehensive review that emphasizes key considerations in target selection, summarizes breakthroughs in the application of AI-driven approaches for therapeutic target exploration, and highlights clinical-stage successes in which #AI played a pivotal role in target identification.
The review, titled "Target identification and assessment in the era of AI", was published in Nature Reviews Drug Discovery (doi.org/10.1038/s41573-026-0…), a leading journal in the pharmaceutical and biomedical research community known for its peer-reviewed strategic roadmaps and in-depth analysis of emerging trends and breakthroughs in drug discovery and development.

4
6
5
1,060
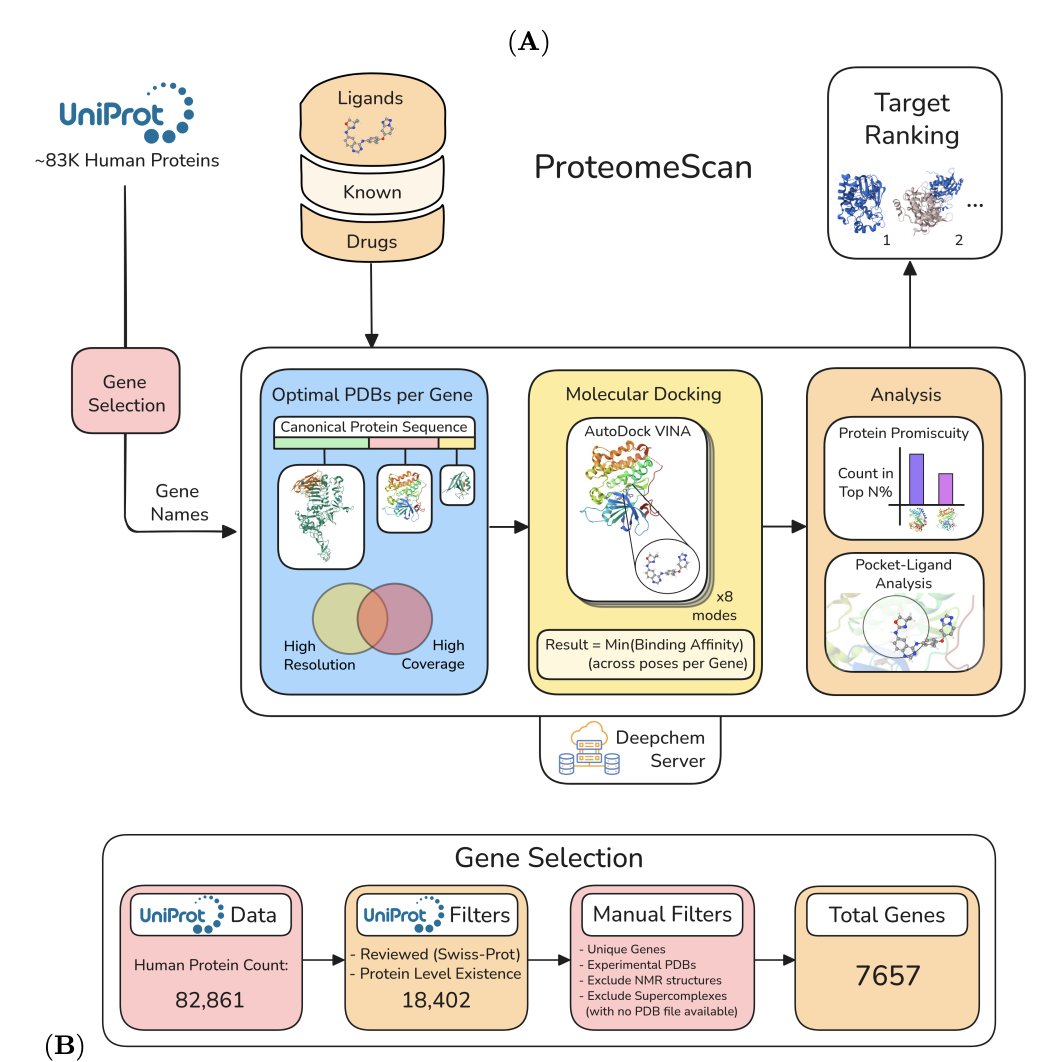
ProteomeScan: A Toolkit For Target Validation By Proteome-Wide Docking And Analysis
1. ProteomeScan is a gene-driven, proteome-wide “one compound–many targets” docking toolkit: it selects an optimal experimental PDB structure set per human gene, runs large-scale blind docking, and ranks candidate targets by predicted binding affinity.
2. Scale and coverage: starting from 82,861 UniProt human protein entries, the authors curate to 7,657 unique genes with experimental PDB structures (excluding NMR models and very large supercomplexes), then dock 20 drug-like ligands across the proteome (>300,000 docking tasks; ~93.65% completion).
3. Key practical innovation: a systematic “protein promiscuity” analysis to identify proteins that repeatedly appear as top hits across many ligands (broad binders that can dominate rankings and inflate false positives). Promiscuity is defined by being in the top m% for at least n of N ligands, enabling tunable filtering.
4. Performance is evaluated with a proteome-coverage metric, Known Target Recovery (KTR): the fraction of known drug–target pairs recovered within the top m% of ranked predictions. ProteomeScan significantly outperforms random rankings (Mann–Whitney U p-values ~1e-4 to 1e-5), and moderate promiscuity filtering improves KTR (e.g., at top 5%: 13.64% baseline ProteomeScan vs 20.45% after removing 166 promiscuous targets).
5. Pose validation is treated as a first-class step, not an afterthought: docking hits are checked for whether ligands actually occupy druggable pockets using fpocket plus custom overlap metrics (e.g., % Ligand Inside Pocket; % Pocket Filled), typically focusing on the top 5–10 pockets by fpocket druggability.
6. Mutant-aware analysis: the study tests whether docking ranks clinically relevant mutant variants above wild type when expected. It succeeds for examples like dabrafenib preferring BRAF V600E over wild-type BRAF, and alpelisib ranking PIK3CA mutants (H1047R, E545K) above wild type—while also highlighting failures where docking scores alone can be misleading without pocket-occupancy checks.
7. The paper explicitly maps where proteome-wide blind docking breaks down: assembly-dependent binding (paclitaxel requiring microtubule assembly; RMC-6236 requiring a tri-complex “molecular glue” mechanism) and cases where favorable scores come from non-druggable surface sites (revealed by low pocket occupancy).
8. Biological interpretation of promiscuous targets: the top promiscuous set (e.g., 166 proteins at top 25% across all 20 ligands) is enriched for families known for broad ligand binding (CYP3A4, GSTs, AKRs, ADH/ALDH enzymes, HSP90), plus potential “over-predicted” large/flexible pockets (e.g., some HDAC/ion channel/GPCR cases) flagged as needing experimental follow-up.
9. A secondary check on promiscuity realism: methylation sensitivity experiments on kinase-like promiscuous targets show methylated ligands often reduce predicted affinity, consistent with pocket-specific interactions; cases with minimal change suggest weaker/non-specific docking interactions.
10. Implementation and accessibility: the workflow is exposed via the DeepChem Server (FastAPI asynchronous jobs), scales via a cloud backend (Prithvi/AWS Batch in the commercial deployment), and the authors emphasize reproducibility by open-sourcing the core algorithm within the DeepChem ecosystem.
💻Code: github.com/deepforestsci/Pro…
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #DrugDiscovery #MolecularDocking #TargetIdentification #ChemicalBiology #Bioinformatics #DeepChem #Proteomics #Polypharmacology #Toxicology
1
3
33
4,409
Apr 14
LAST CHANCE! 🔇
Please join us today April 14th at 10 AM ET, for our Pharma.AI Spring Kickoff.
🐼 We’ll be walking through our latest PandaOmics updates, with a chance to see #PandaClaw, our upcoming agentic AI tool in action, and discover how its natural-language interface allows you to conduct complex, real-time multi-omics analyses and generate research hypotheses effortlessly.
We will also demonstrate how our recent incorporation of comprehensive single-cell datasets provides enhanced resolution for your #targetidentification.
You can register here [insilico.zoom.us/webinar/reg…] to secure your spot for the session.
If you aren't able to join us live, make sure you register and be the first to receive the on-demand recording to watch at your convenience.
#PharmaAI2026 #Q1Update #PharmaAIWebinar
1
2
3
556
Mar 18
🧬 PandaOmics Wednesday Target Challenge is LIVE!
Can you guess this week’s target?
🔍 Clues:
• Activated during tissue stress on epithelial surfaces
• Plays a key role in TGF-β signaling
• Implicated in cancer progression and fibrosis
• A promising druggable switch (without blocking TGF-β entirely)
💡 This target sits at the intersection of tumor invasion, fibrosis, and microenvironment remodeling, making it a highly attractive (and nuanced) therapeutic opportunity.
👇 Drop your guess below.
We’ll reveal the answer next week.
#PandaOmics #DrugDiscovery #TargetIdentification #AI #Biotech #ComputationalBiology #PrecisionMedicine
3
6
350

📢New Research Topic published in the Antimicrobials, Resistance and Chemotherapy section!! 📢
AI-Driven Antimicrobial Drug Discovery
📚 Join Frontiers in Microbiology in this exploration to advance the understanding on AI-based approaches to tackle Antimicrobial Resistance.
Learn more and submit your work here: fro.ntiers.in/RT77380
For questions, please contact us at microbiology@frontiersin.org
#AntimicrobialResistance #ArtificialIntelligence #MachineLearning #DrugDiscovery #TargetIdentification #AntimicrobialResistanceSurveillance

2
2
157
Feb 10
Day 5 of #ScienceAIBench: Target Identification 🎯
We continue our daily release series with Day 5, shifting focus from molecular properties to the complex challenge of biological target discovery. Identifying the right therapeutic target is the most critical step in drug development to avoid costly clinical failures. Today, we dig deeper into bioinformatics and introduce the results for the TargetBench module.
Success in drug discovery begins with the right target. Today's benchmark evaluates the ability of frontier AI models to recover established, clinical-stage targets for complex fibrotic diseases, distinguishing true biological signals from noise.
We have also published a preprint detailing our methodology and full results, available now alongside the live benchmark data on our website.
📄 Read the Preprint: [biorxiv.org/content/10.1101/…] 🌐 View the Benchmark: [targetbench.org/]
📋 Benchmark Specifications:
Disease Indications: Idiopathic Pulmonary Fibrosis (IPF), Atherosclerosis, and Osteoarthritis.
Metric: Clinical Target Recall (CTR) — the proportion of known clinical-stage targets retrieved in the model's top-ranked predictions.
Models Evaluated: GPT-5, Claude Opus 4, Grok 4, and DeepSeek R1.
📊 Observed Performance:
Top Performer: GPT-5 demonstrated the highest Clinical Target Recall across all tested indications, achieving 0.474 for Atherosclerosis and 0.352 for IPF.
Performance Stratification: A distinct gap exists between models; Claude Opus 4 ranked second (e.g., 0.377 for Atherosclerosis), while Grok 4 and DeepSeek R1 generally showed lower retrieval rates.
Complex Disease Challenge: IPF proved to be the most difficult indication for baselines, with Grok 4 recording its lowest score of 0.199, highlighting the difficulty of reasoning over complex fibrotic pathology.
General Trend: The data reveals significant stratification in reasoning capabilities for biological target discovery, with clear performance tiers between model generations.
🔄 An updated version of TargetBench with expanded disease indications is coming soon. Stay tuned for further insights.
#ScienceAI #InsilicoBench #DrugDiscovery #AI #TargetIdentification #Bioinformatics #MachineLearning

2
3
8
773

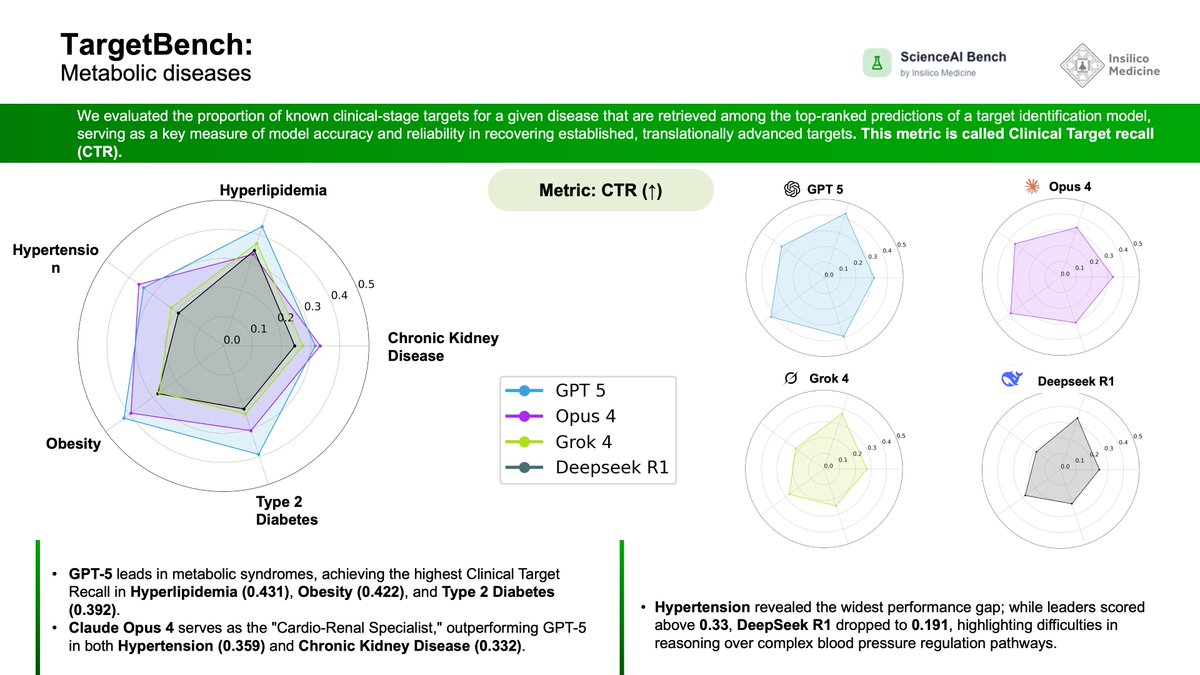
Day 6 of #ScienceAIBench! 🧬 Following our exploration of fibrotic diseases yesterday, we are shifting focus to systemic pathology. Today’s update releases the Metabolic module of TargetBench, testing how well frontier models can identify clinical-stage targets for high-prevalence conditions like Type 2 Diabetes and Hypertension.
We have published a preprint detailing our methodology and full results, available now alongside the live benchmark data on our website.
📄 Read the Preprint: biorxiv.org/content/10.1101/…
🌐 View the Benchmark: targetbench.org/
📋 Benchmark Specifications:
Disease Indications: Chronic Kidney Disease (CKD), Hyperlipidemia, Hypertension, Obesity, and Type 2 Diabetes (T2D).
Metric: Clinical Target Recall (CTR) — the proportion of known clinical-stage targets retrieved in the model's top predictions.
Models Evaluated: GPT-5, Claude Opus 4, Grok 4, and DeepSeek R1.
📊 Observed Performance:
Best Performer (Metabolic Syndromes): GPT-5 secured the highest recall in conditions associated with systemic metabolism, leading in Hyperlipidemia (0.431), Obesity (0.422), and Type 2 Diabetes (0.392).
Cardio-Renal Specialization: Claude Opus 4 was better in indications involving hemodynamics and renal function, achieving top scores for Hypertension (0.359) and Chronic Kidney Disease (0.332).
Reasoning Gaps: Hypertension proved to be the most discriminatory task; while leading models scored above 0.33, DeepSeek R1 dropped to 0.191, highlighting difficulties in reasoning over complex blood pressure regulation pathways.
🔄 Our daily benchmark series continues tomorrow.
#ScienceAI #InsilicoBench #DrugDiscovery #AI #MetabolicDisease #TargetIdentification #Bioinformatics #Type2Diabetes
1
1
8
1,131
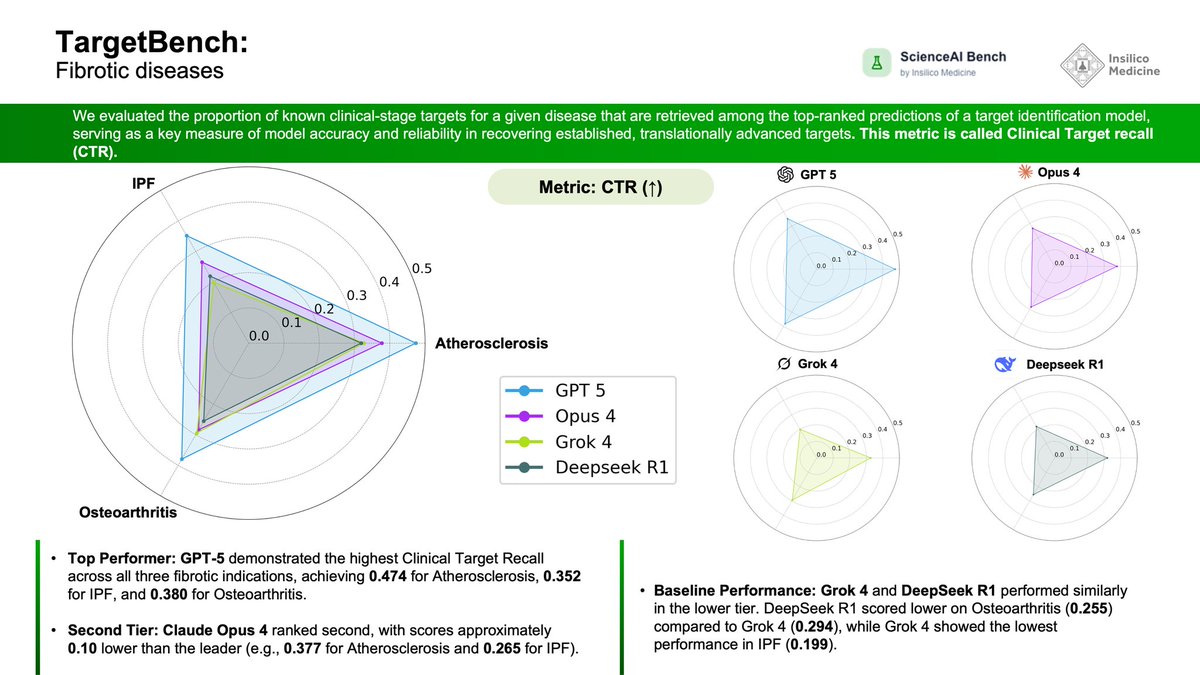
Day 5 of #ScienceAIBench: Target Identification 🎯
We continue our daily release series with Day 5, shifting focus from molecular properties to the complex challenge of biological target discovery. Identifying the right therapeutic target is the most critical step in drug development to avoid costly clinical failures. Today, we dig deeper into bioinformatics and introduce the results for the TargetBench module.
Success in drug discovery begins with the right target. Today's benchmark evaluates the ability of frontier AI models to recover established, clinical-stage targets for complex fibrotic diseases, distinguishing true biological signals from noise.
We have also published a preprint detailing our methodology and full results, available now alongside the live benchmark data on our website.
📄 Read the Preprint: [biorxiv.org/content/10.1101/…] 🌐 View the Benchmark: [targetbench.org/]
📋 Benchmark Specifications:
Disease Indications: Idiopathic Pulmonary Fibrosis (IPF), Atherosclerosis, and Osteoarthritis.
Metric: Clinical Target Recall (CTR) — the proportion of known clinical-stage targets retrieved in the model's top-ranked predictions.
Models Evaluated: GPT-5, Claude Opus 4, Grok 4, and DeepSeek R1.
📊 Observed Performance:
Top Performer: GPT-5 demonstrated the highest Clinical Target Recall across all tested indications, achieving 0.474 for Atherosclerosis and 0.352 for IPF.
Performance Stratification: A distinct gap exists between models; Claude Opus 4 ranked second (e.g., 0.377 for Atherosclerosis), while Grok 4 and DeepSeek R1 generally showed lower retrieval rates.
Complex Disease Challenge: IPF proved to be the most difficult indication for baselines, with Grok 4 recording its lowest score of 0.199, highlighting the difficulty of reasoning over complex fibrotic pathology.
General Trend: The data reveals significant stratification in reasoning capabilities for biological target discovery, with clear performance tiers between model generations.
🔄 An updated version of TargetBench with expanded disease indications is coming soon. Stay tuned for further insights.
#ScienceAI #InsilicoBench #DrugDiscovery #AI #TargetIdentification #Bioinformatics #MachineLearning
2
2
16
3,995

11 Oct 2025
SMR - Final Call to Register for the Target Identification and Validation in Drug Discovery Meeting
@SocMedRes @AstraZeneca
oncodaily.com/voices/drug-di…
#Cancer #Medicine #Health #Oncology #OncoDaily #DrugDiscovery #TargetIdentification #SMR #MedX #MedNews #MedEd

1
233

9 Jun 2025
Our CTO envisions a future where large-scale data and virtual cell models drive #targetidentification and #drugdiscovery. Get his take on what’s needed to leverage the full potential of #singlecell and #spatial data for AI-driven therapeutic development: bit.ly/456TC2e

7
13
974

9 Jun 2025
📢 We are excited to announce OriGene, a self-evolving virtual disease biologist poised to revolutionize therapeutic target discovery. This AI agent system autonomously discovers novel, mechanistically grounded drug targets at scale, addressing the critical bottleneck in drug development🧬
Key features:
🔥Cognitive architecture: Employs a multi-agent system mimicking expert reasoning with "Thinking Templates" to mitigate "logical hallucinations."
🔥Adaptive tooling: Dynamically integrates over 500 specialized tools and databases, crafting bespoke solutions for complex biological queries.
🔥Continuous learning: Features a two-level self-evolution mechanism, enabling self-correction from experimental feedback without costly retraining.
Our latest validations confirm the discovery and wet-lab validation of ARG2 for colorectal cancer and GPR160 for liver cancer as novel therapeutic targets. OriGene's superior performance is evident: it outscored human experts 60.08% to 55% on the curated TRQA benchmark, demonstrated up to 32.1% improvement over leading LLMs like GPT-4o, and achieved top results across GPQA, DbQA, and LitQA, showcasing its powerful scientific reasoning.
OriGene is an open-source project! We're actively polishing the code for an upcoming release and warmly invite the community to build on it, joining us to advance the future of medicine.
🛠️ Code: github.com/GENTEL-lab/OriGen…
📄Paper: biorxiv.org/content/10.1101/…
🧬 Project Page: gentel-lab.github.io/OriGene…
📊 Benchmark: huggingface.co/datasets/GENT…
#AIforDrugDiscovery #AgenticAI #GenerativeAI #TargetIdentification #BiomedicalResearch #LLMs
2
10
671

2 Jun 2025
RAGPPI: RAG Benchmark for Protein-Protein Interactions in Drug Discovery
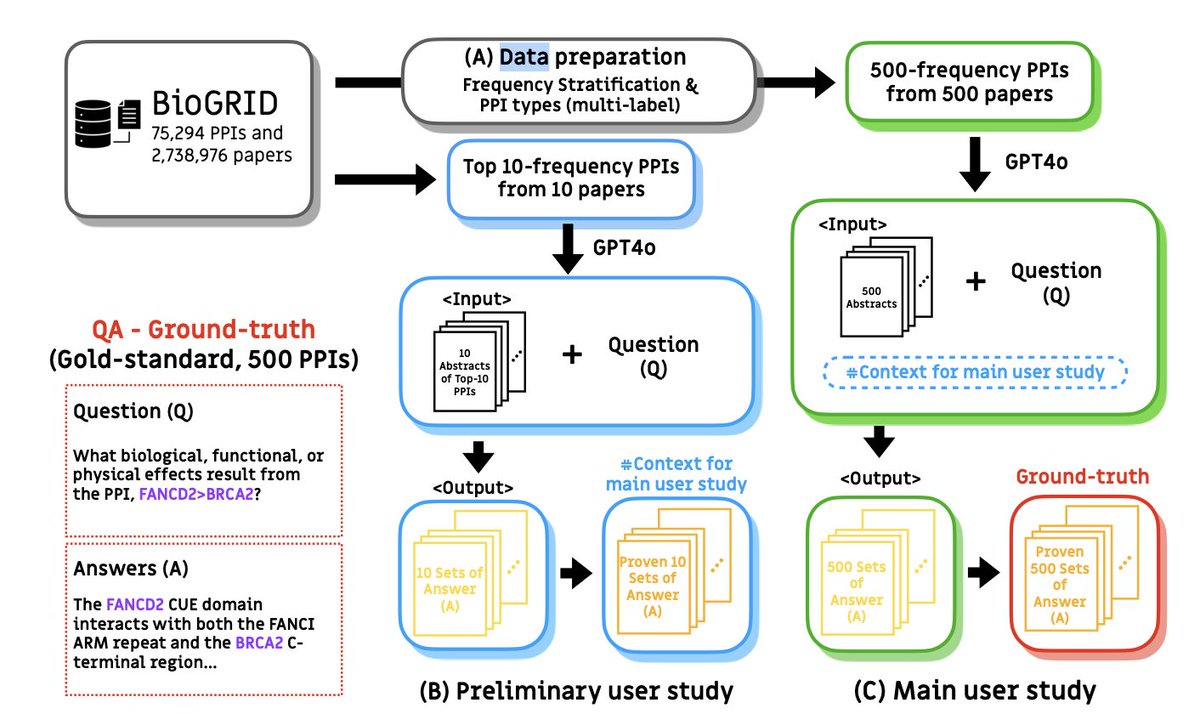
1.RAGPPI introduces the first benchmark specifically designed to evaluate the ability of Retrieval-Augmented Generation (RAG) systems to reason about the biological impacts of protein-protein interactions (PPIs), a key step in drug target identification.
2.At its core, RAGPPI provides a factual QA benchmark consisting of 4,420 question-answer pairs. These questions probe how specific PPIs affect biological or therapeutic outcomes, reflecting the real-world needs of drug discovery researchers.
3.The benchmark includes a high-quality gold-standard subset of 500 QA pairs, validated and revised by ten domain experts through rigorous user studies. These experts annotated LLM-generated answers for accuracy and completeness.
4.To scale the dataset, the authors developed an ensemble auto-evaluation model composed of three GPT-4o-based evaluators. These models emulate expert judgments by analyzing atomic fact similarity to ground-truth abstracts using cosine similarity metrics.
5.The ensemble model achieves 93.71% accuracy and enables the creation of a silver-standard set of 3,720 QA pairs. This approach provides large-scale annotated data without requiring additional expert labor.
6.Expert interviews guided the design of the benchmark. Researchers emphasized the need for evaluating downstream biological effects of PPIs using balanced reference distributions across interaction types and literature frequencies.
7.The authors structured the benchmark to cover seven PPI types (e.g., Binding, Activation, Inhibition) and three literature frequency levels (high, medium, low), ensuring comprehensive coverage across common and rare PPIs.
8.They constructed the gold-standard dataset using BioGRID, selecting PPIs from curated abstracts and maintaining balance in both type and frequency. This ensured that evaluation contexts were realistic and diverse.
9.Evaluation experiments showed that general-purpose LLMs like Gemini-2.0-Flash outperformed existing RAG pipelines such as GraPPI and GeneGPT on factual accuracy. Surprisingly, current RAG models struggled due to weak retrievers and hallucinations.
10.The study underscores that high semantic similarity (e.g., BERTScore) does not equate to factual correctness. The ensemble evaluator proved better aligned with expert assessments than traditional LLMs or semantic metrics alone.
11.To encourage community adoption, the authors release not just the dataset but also the ensemble auto-evaluator. This allows reproducible benchmarking and facilitates future RAG system development for biomedical QA.
12.Limitations include the relatively small size of the gold-standard set and the limited number of domain experts involved. Future work aims to expand annotations and apply RAGPPI to broader drug discovery tasks like ligand design and repurposing.
💻Code: github.com/youngseungjeon/RA… 📜Paper: arxiv.org/abs/2505.23823v1
#DrugDiscovery #ProteinInteractions #LLM #RAG #BiomedicalAI #Bioinformatics #TargetIdentification
3
2
910
2 Jun 2025
RAGPPI: RAG Benchmark for Protein-Protein Interactions in Drug Discovery
1.RAGPPI introduces the first benchmark specifically designed to evaluate the ability of Retrieval-Augmented Generation (RAG) systems to reason about the biological impacts of protein-protein interactions (PPIs), a key step in drug target identification.
2.At its core, RAGPPI provides a factual QA benchmark consisting of 4,420 question-answer pairs. These questions probe how specific PPIs affect biological or therapeutic outcomes, reflecting the real-world needs of drug discovery researchers.
3.The benchmark includes a high-quality gold-standard subset of 500 QA pairs, validated and revised by ten domain experts through rigorous user studies. These experts annotated LLM-generated answers for accuracy and completeness.
4.To scale the dataset, the authors developed an ensemble auto-evaluation model composed of three GPT-4o-based evaluators. These models emulate expert judgments by analyzing atomic fact similarity to ground-truth abstracts using cosine similarity metrics.
5.The ensemble model achieves 93.71% accuracy and enables the creation of a silver-standard set of 3,720 QA pairs. This approach provides large-scale annotated data without requiring additional expert labor.
6.Expert interviews guided the design of the benchmark. Researchers emphasized the need for evaluating downstream biological effects of PPIs using balanced reference distributions across interaction types and literature frequencies.
7.The authors structured the benchmark to cover seven PPI types (e.g., Binding, Activation, Inhibition) and three literature frequency levels (high, medium, low), ensuring comprehensive coverage across common and rare PPIs.
8.They constructed the gold-standard dataset using BioGRID, selecting PPIs from curated abstracts and maintaining balance in both type and frequency. This ensured that evaluation contexts were realistic and diverse.
9.Evaluation experiments showed that general-purpose LLMs like Gemini-2.0-Flash outperformed existing RAG pipelines such as GraPPI and GeneGPT on factual accuracy. Surprisingly, current RAG models struggled due to weak retrievers and hallucinations.
10.The study underscores that high semantic similarity (e.g., BERTScore) does not equate to factual correctness. The ensemble evaluator proved better aligned with expert assessments than traditional LLMs or semantic metrics alone.
11.To encourage community adoption, the authors release not just the dataset but also the ensemble auto-evaluator. This allows reproducible benchmarking and facilitates future RAG system development for biomedical QA.
12.Limitations include the relatively small size of the gold-standard set and the limited number of domain experts involved. Future work aims to expand annotations and apply RAGPPI to broader drug discovery tasks like ligand design and repurposing.
💻Code: github.com/youngseungjeon/RA…
📜Paper: arxiv.org/abs/2505.23823v1
#DrugDiscovery #ProteinInteractions #LLM #RAG #BiomedicalAI #Bioinformatics #TargetIdentification

1
6
1,000

24 Oct 2024
2/3 We work on #GenomeStability #DNARepair #TargetIdentification #TargetValidation using genomics, proteomics and chemical biology in #CancerResearch.
We are looking for pre-PhDs, postdocs and a lab manager.
1
1
8
647

30 Apr 2024
At BenevolentAI, we’re proud of our Target Identification offering. It has yielded great results for partners, as well as for our own pipeline.
🎯 Quickly predict and evaluate the relevance, novelty and progressability of potential drug targets.
#AI #TargetIdentification

8
1,116

25 Mar 2024
Following our successful work with @sanofi on #targetidentification, we’re proud to announce the next phase of the collaboration, focused on drug positioning within Sanofi’s immunology therapeutic pipeline.
Owkin will combine existing knowledge and cutting-edge #machinelearning to optimize #drugpositioning and match drugs with the most effective disease indications and subpopulations, ultimately enhancing patient outcomes.
Read more at owkin.com/newsfeed/owkin-exp…
Learn more about our AI-driven approach to optimize clinical development owkin.com/drug-development-a…
2
3
2,013

18 Jan 2024
Stop by the Wilcox Booth 62303 next week at #SHOTSHOW to learn more about the RAPTAR Xe.
@blueforcedev #BTAC
#SHOTSHOW2024 #RangeFinder #RapidTargeting #Sniper #NIRIllumination #HighPowerLaser #BallisiticSolution #TargetIdentification #RAPTARS #RAPTARXe

1
4
339

27 Sep 2023
Yesterday, we celebrated the 25th anniversary at Hybrigenics for 3 of us. Congratulations to Céline, Véronique and @JChristopheRain .
All our warm thanks for your achievements. So Many #Y2H screens, #ProteinInteractions, #TargetIdentification, #InducedProximity & more recently selection and optimization of #Nanobodies #AnimalFree #TechnologyPlatform

4
7
330