
Learning Structure, Energy, and Dynamics: A Survey of Artificial Intelligence for Protein Dynamics
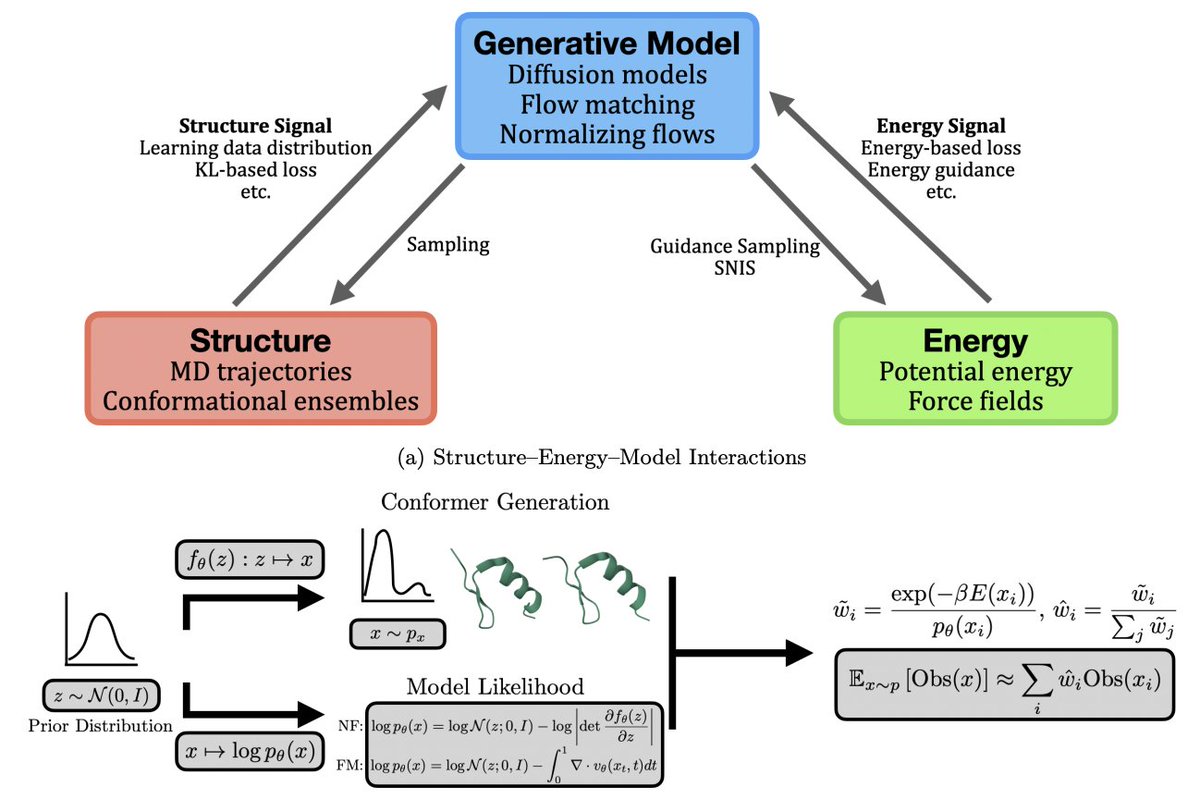
1. The survey maps the protein-dynamics AI landscape into three complementary training signals: learning from structural ensembles/trajectories, learning from physical energy signals (Boltzmann learning), and learning to accelerate or replace components of molecular dynamics (ML force fields, coarse graining, and collective variables).
2. A key theme is the shift from inference-time “make AlphaFold diverse” heuristics to explicit generative modeling of p(x|sequence) for equilibrium ensembles, using diffusion, flow matching, and latent language-modeling approaches that can be trained end-to-end on ensemble data and sampled efficiently.
3. For equilibrium ensemble generation, it highlights how modern methods incorporate stronger priors and broader conditioning: MSA-free sequence-conditioned generators (e.g., latent diffusion with protein language models), temperature/thermodynamic conditioning, and energy-conditioned sampling to target distinct conformational states rather than a single dominant fold.
4. It emphasizes practical failure modes in purely data-driven ensemble learning—limited physical realism, scarcity of diverse dynamic data, and PDB conformational bias—and surveys mitigation strategies such as force/energy guidance, physics-informed objectives (e.g., Fokker–Planck supervision), experimental stability signals, and dataset reweighting via structural clustering.
5. The review extends “dynamics generation” beyond i.i.d. conformers to explicit trajectory models p(X|x1): (i) learned long-timestep transition kernels (MCMC-style), (ii) autoregressive frame prediction with improved temporal scalability (including state-space-model adaptations), and (iii) one-shot “trajectory-as-video” generation that outputs full time-ordered coordinate sequences in a single pass.
6. A central innovation thread is energy-driven learning: Boltzmann generators and related samplers that use energies/forces to learn proposals for the Boltzmann distribution, then correct residual bias with self-normalized importance sampling or annealing/SMC-style procedures, with effective sample size (ESS) used as a key reliability diagnostic.
7. The survey contrasts exact-likelihood normalizing-flow Boltzmann generators (enabling principled reweighting) with likelihood-free diffusion/flow approaches trained from energetic supervision, and discusses the core tradeoff: thermodynamic guarantees vs scalability, symmetry handling, and computational cost of likelihood/energy evaluations.
8. It also covers “physics-aware adaptation” of pretrained protein generators: post-training alignment and inference-time steering that tilt a base generator toward lower-free-energy or constraint-satisfying samples using energies, forces, projections, or CV/observable-based biases—aiming to retrofit thermodynamic meaning without retraining from scratch.
9. On the simulation side, it reviews how ML accelerates or upgrades MD via: (i) machine-learning potentials approaching QM fidelity while scaling to large biomolecular systems (often via fragmentation, Δ-learning, and explicit long-range terms), (ii) learned coarse-grained models that approximate potentials of mean force for longer timescales, and (iii) ML-derived collective variables for enhanced sampling using dimensionality reduction, kinetic learning, RL/adaptive sampling, and differentiable generative constraints.
10. The survey closes by curating key datasets (static PDB and distilled AFDB/AFESM; MD trajectory corpora like ATLAS, DynamicPDB, mdCATH, MISATO, DD-13M; and IDP/experimental resources like BMRB and SASBDB) and framing open challenges: scalability, thermodynamic consistency, kinetic fidelity, dataset/force-field bias, and principled integration with experimental constraints.
📜Paper: arxiv.org/abs/2604.25244
#ProteinDynamics #ComputationalBiology #MolecularDynamics #GenerativeAI #DiffusionModels #FlowMatching #BoltzmannGenerators #MachineLearningPotentials #CoarseGraining #EnhancedSampling
1
4
40
2,437
Impact of Local Descriptors Derived from Machine Learning Potentials in Graph Neural Networks for Molecular Property Prediction
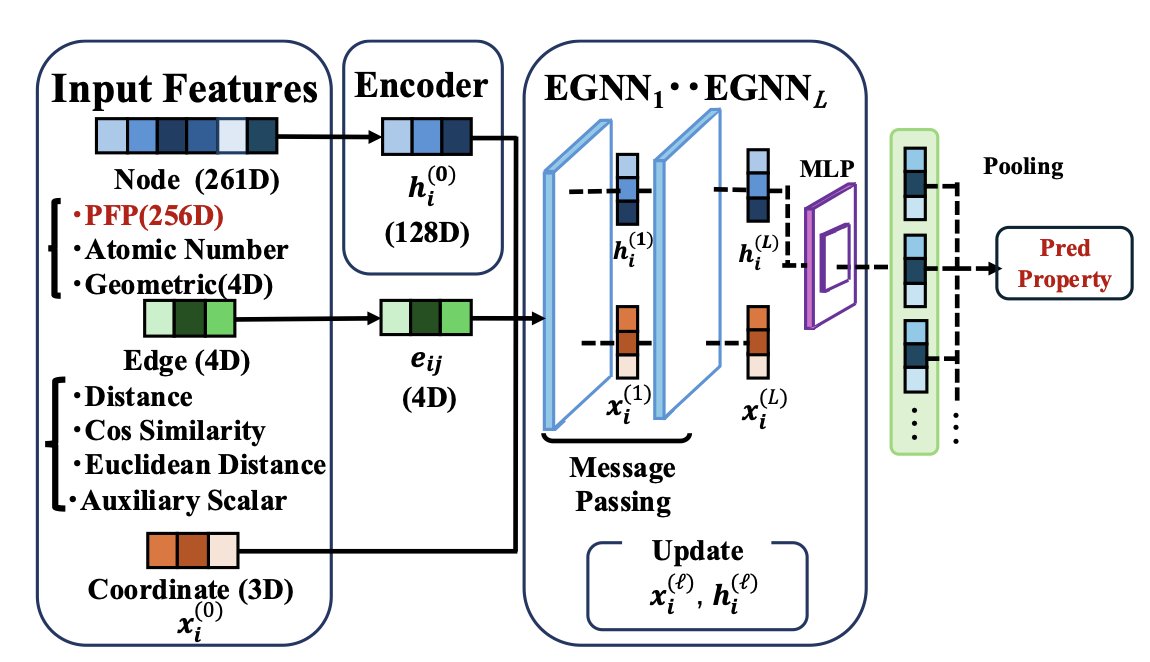
1 The authors propose a framework that enhances molecular property prediction by integrating pretrained local descriptors from machine learning potentials into 3D graph neural networks, demonstrating that local electronic environment information significantly improves prediction accuracy beyond geometric features alone.
2 The core innovation lies in using 256-dimensional PFP (Preferred Potential) descriptors extracted from Matlantis as node features in an EGNN architecture, where these descriptors encode rich local atomic environment information learned from large-scale DFT calculations.
3 The EGNN-PFP model was evaluated on two distinct datasets: QM9 (133,831 organic molecules with 12 properties) and tmQM (86,000 transition metal complexes with 5 properties), showing consistent improvements across diverse chemical spaces.
4 For the QM9 dataset, the model achieved superior accuracy compared to both original EGNN and baseline models for 11 out of 12 properties, with notable error reductions of approximately 24% for dipole moment, 21% for HOMO energy, and 15% for HOMO-LUMO gap.
5 The most dramatic improvements were observed on the tmQM dataset containing transition metal complexes, where error reductions reached approximately 63% for dipole moment, 52% for HOMO-LUMO gap, 44% for HOMO energy, 43% for LUMO energy, and 65% for metal partial charge.
6 The study reveals that pretrained local descriptors are particularly effective for transition metal systems with complex d-orbital electronic structures, where traditional geometric-only approaches struggle to capture the intricate electronic environments around metal centers.
7 A key advantage of this approach is its element-agnostic nature: the same model architecture works effectively across vastly different chemical spaces, from 5-element organic molecules (H, C, N, O, F) to 30 element transition metal complexes, without requiring retraining or architectural modifications.
8 The method is computationally efficient as PFP descriptors are obtained through a simple forward pass of the pretrained potential, eliminating the need for additional quantum chemical calculations that methods like NatQG (NBO analysis) or QTAIM-GNN (QTAIM analysis) require.
9 The authors emphasize that their contribution is not about improving a specific GNN architecture, but rather establishing a general design principle: enriching node features with pretrained local electronic representations can systematically enhance molecular property prediction across various 3D GNN frameworks.
10 The framework demonstrates how machine learning potentials, traditionally focused on energy and force predictions, can be leveraged to significantly expand the scope of accurately predictable molecular properties through their internal representations.
📜Paper: arxiv.org/abs/2502.03046
#MolecularPropertyPrediction #GraphNeuralNetworks #MachineLearningPotentials #ComputationalChemistry #DeepLearning #QuantumChemistry #TransitionMetalComplexes #Matlantis #EGNN #ChemInformatics
1
5
1,080

20 Sep 2023
Still time to register for tomorrow's webinar on the new Synopsys @QuantumATK V-2023.09 release 👉
bit.ly/4698CcW
#atomisticsimulation #materialsscience #semiconductorindustry #machinelearningpotentials #dft
12 Sep 2023

We are excited to announce the new @QuantumATK V-2023.09 release. Join our free @Synopsys Webinar on Sep 21 to learn about the new features and improvements for many atomic-scale modeling methods and applications in semiconductor industry and beyond: bit.ly/4698CcW

2
166