
PhD student at MIT Micro. Genomic language models, Metagenomics, phage enthusiast, Dead Head.
Joined January 2022
- Tweets 111
- Following 615
- Followers 225
- Likes 13,975
5 Photos and videos





Jordan Hoff retweeted

Jun 8
Max-pooling sparse features > mean-pooling dense features
Pooling nerds will enjoy this part of the ESMC interpretability work.
Protein language models have to capture *everything* about a sequence, including the organism that the sequence came from, its structural fold, etc in order to be able predict amino acids.
If a functional signal (e.g. active site, allostery) is local in sequence space, it may be drowned out by mean-pooling.
Max-pooling across sparse features preserves this signal, and we show that it improves functional homolog recovery in the presence of decoys.

Jun 5

Furthermore, if we maxpool the features across the sequence dimension, we reduce the effect from low frequency averaging of dense embeddings, and get a vocabulary of strongly activated functional features for each protein.
In fact, now we can use some of the common techniques found in information retrieval, such as TF-IDF and BM25 [our Jaccard similarity is close to this] for protein search.
2
10
65
4,136
Jordan Hoff retweeted

May 21
🚀 Excited to share our new work: Absolute Stability Predictor!
📊: forms.gle/4ZnXZSnTBvaykkAi9
Built the MGnify Stability Dataset (1.8M measurements) and developed stability prediction models, together with @grocklin, @KotaroTsuboyama, @sokrypton, and teams.
5
62
231
39,072
Jordan Hoff retweeted

Apr 30
1/ Excited to share our new paper in Science: “Toward life with a 19-amino acid alphabet through generative artificial intelligence design.” @ColumbiaSysBio @ColumbiaBME @Columbia
science.org/doi/10.1126/scie… 🦠🧬🛠️🖥️💥
17
155
542
84,559
Jordan Hoff retweeted

Apr 27
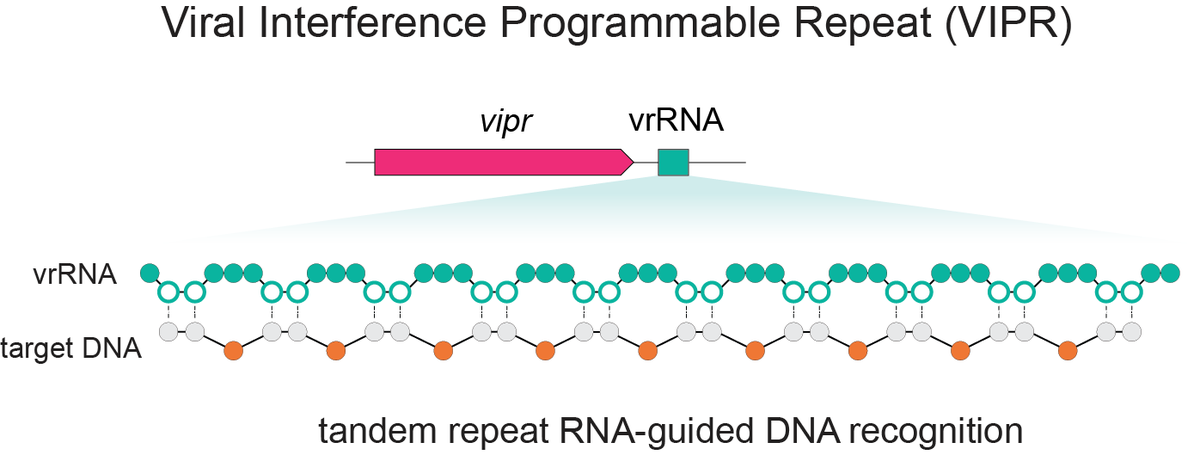
Excited to share our discovery of a new programmable RNA-guided DNA-targeting system hiding inside bacteriophages that predates CRISPR.
We call it VIPR (Viral Interference Programmable Repeat), and it uses an entirely new logic to find its targets.
Thread link below.
26
353
1,257
241,134
Jordan Hoff retweeted

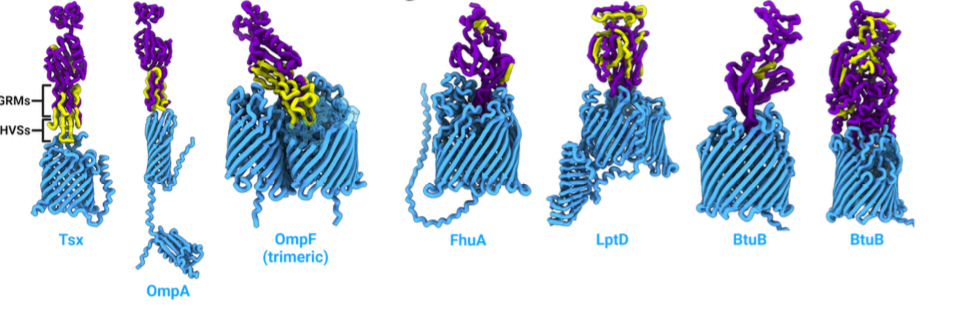
🥳 Huge preprint 🔔 Today we share something our group has been working toward for a long time, led by
@LucasMoriniere We asked can we predict which receptor a phage targets from its genome sequence alone? For most phages, we couldn’t. So Lucas set out to do something crazy!
4
66
235
20,802
Jordan Hoff retweeted

Big news! Starting my lab as Ramón y Cajal PI at @IBVF_Sevilla in Seville 🌞, Spain, bridging microbial ecology, photosynthesis & plant biotech: from metagenomic and AI analyses 💻 to experiments in microbes & plants 🌊🧬🌱 Looking for PhD students & postdocs. DM or share!
10
35
109
9,309
Jordan Hoff retweeted

Happy to see this project that started during my PhD come to completion! Beautiful work by @AlexanderRotsch and team solving the structure of the influenza polymerase/elongating RNA polymerase II complex. Congrats to everyone for the amazing story! nature.com/articles/s41586-0…
2
3
24
1,412
Jordan Hoff retweeted

Mar 3
Predicting protein-protein interactions (PPIs) at proteome scale can take months with co-folding models due to massive all-vs-all comparisons required.
We are excited to announce FlashPPI, a contrastive model that predicts proteome wide physical interfaces in minutes. 1/🧵
5
30
131
10,704
Jordan Hoff retweeted

Feb 23
We identify conjugative megaplasmids in the human gut that embed intact temperate prophage genomes and may enable cross-lineage viral dissemination.
We propose a plasmid-mediated route for temperate phage spread.
biorxiv.org/cgi/content/shor…
1
3
3
261
Jordan Hoff retweeted

Jan 12
Introducing DroPE: Extending the Context of Pretrained LLMs by Dropping Their Positional Embeddings
pub.sakana.ai/DroPE/
We are releasing a new method called DroPE to extend the context length of pretrained LLMs without the massive compute costs usually associated with long-context fine-tuning.
The core insight of this work challenges a fundamental assumption in Transformer architecture. We discovered that explicit positional embeddings like RoPE are critical for training convergence but eventually become the primary bottleneck preventing models from generalizing to longer sequences.
Our solution is radically simple: We treat positional embeddings as a temporary training scaffold rather than a permanent architectural necessity.
Real-world workflows like reviewing massive code diffs or analyzing legal contracts require context windows that break standard pretrained models. While models without positional embeddings (NoPE) generalize better to these unseen lengths, they are notoriously unstable to train from scratch.
Here, we achieve the best of both worlds by using embeddings to ensure stability during pretraining and then dropping them to unlock length extrapolation during inference. Our approach unlocks seamless zero-shot context extension without any expensive long-context training.
We demonstrated this on a range of off-the-shelf open-source LLMs. In our tests, recalibrating any model with DroPE requires less than 1% of the original pretraining budget, yet it significantly outperforms established methods on challenging benchmarks like LongBench and RULER.
We have released the code and the full paper to encourage the community to rethink the role of positional encodings in modern LLMs.
Paper: arxiv.org/abs/2512.12167
Code: github.com/SakanaAI/DroPE
39
260
1,777
470,871
Jordan Hoff retweeted

10 Dec 2025
I'm super excited to announce the first preprint of my PhD, together with Chenxi Ou and
@sokrypton!
ML has revolutionized protein modeling, but key challenges remain. For example, we can't predict complicated protein structures without MSAs, which limits what we can design.
8
44
182
76,159
Jordan Hoff retweeted

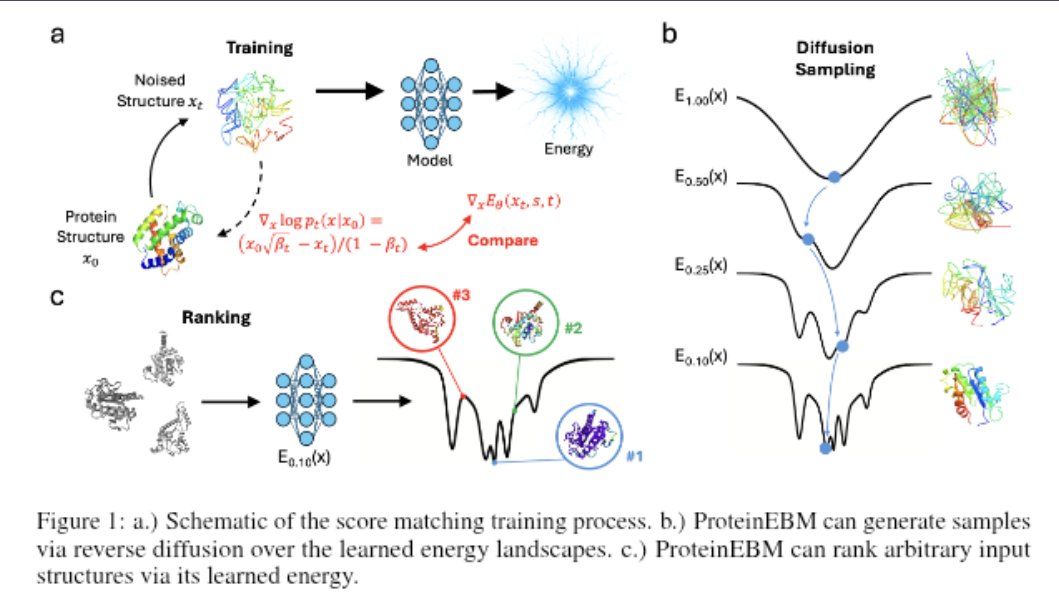
10 Dec 2025
An energy-based model of protein conformational space can be used to predict structure from sequence, sample from the conformational landscape, rank structures, and predict mutation effects.
@jamesproney @sokrypton
3
35
177
9,330
Jordan Hoff retweeted

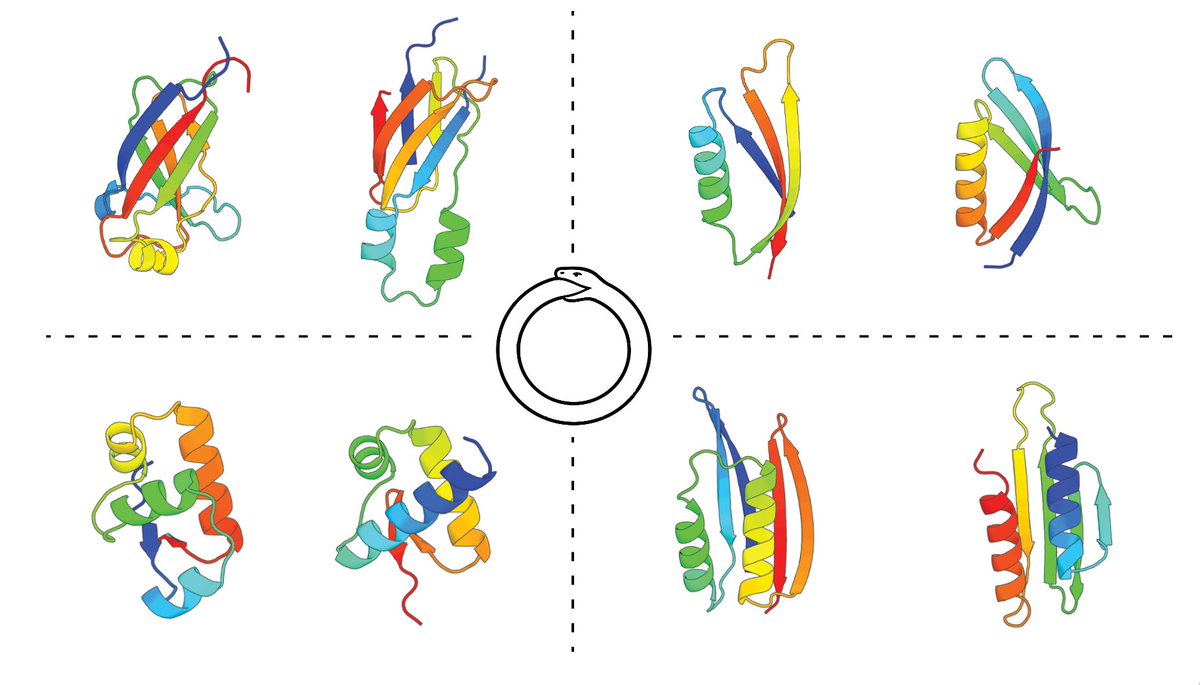
3 Dec 2025
Excited to share my recent work, CIRPIN 🐍 with @sokrypton and @MazAbulnaga on the discovery of thousands of naturally occurring circularly permuted protein domains
5
25
99
16,560
Jordan Hoff retweeted

1 Dec 2025
Excited to share this work with @yoakiyama @ChoYehlin @jajoosam @sokrypton
We find that protein language models trained solely on individual protein sequences, implicitly learn the interface contacts of homo-oligomeric assemblies! As the model scales up, more interface signals pop up.
However, we also notice some proteins PDB database marked as homo-oligomers are missing these interface signals. This took us on the journey to investigate the emergence and absence of homo-oligomeric contacts in pLMs.
biorxiv.org/content/10.1101/…
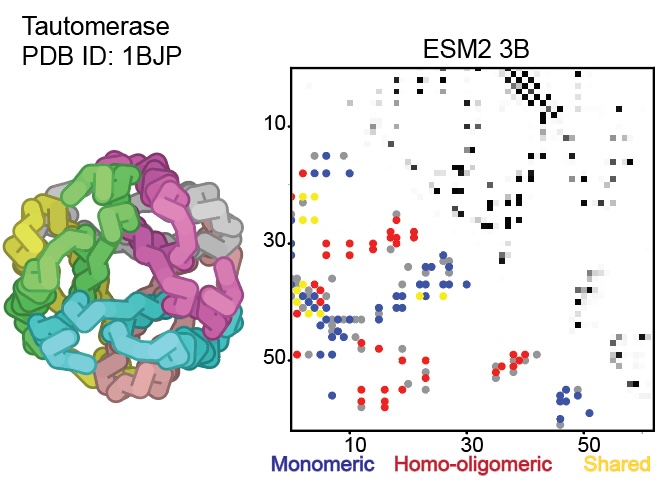
4
30
139
13,152
Jordan Hoff retweeted

29 Oct 2025
Is 3D dragging you down? Wish you could instead use the 2D ColabFold representation for all your work? 🤓
Introducing: py2Dmol 🧬
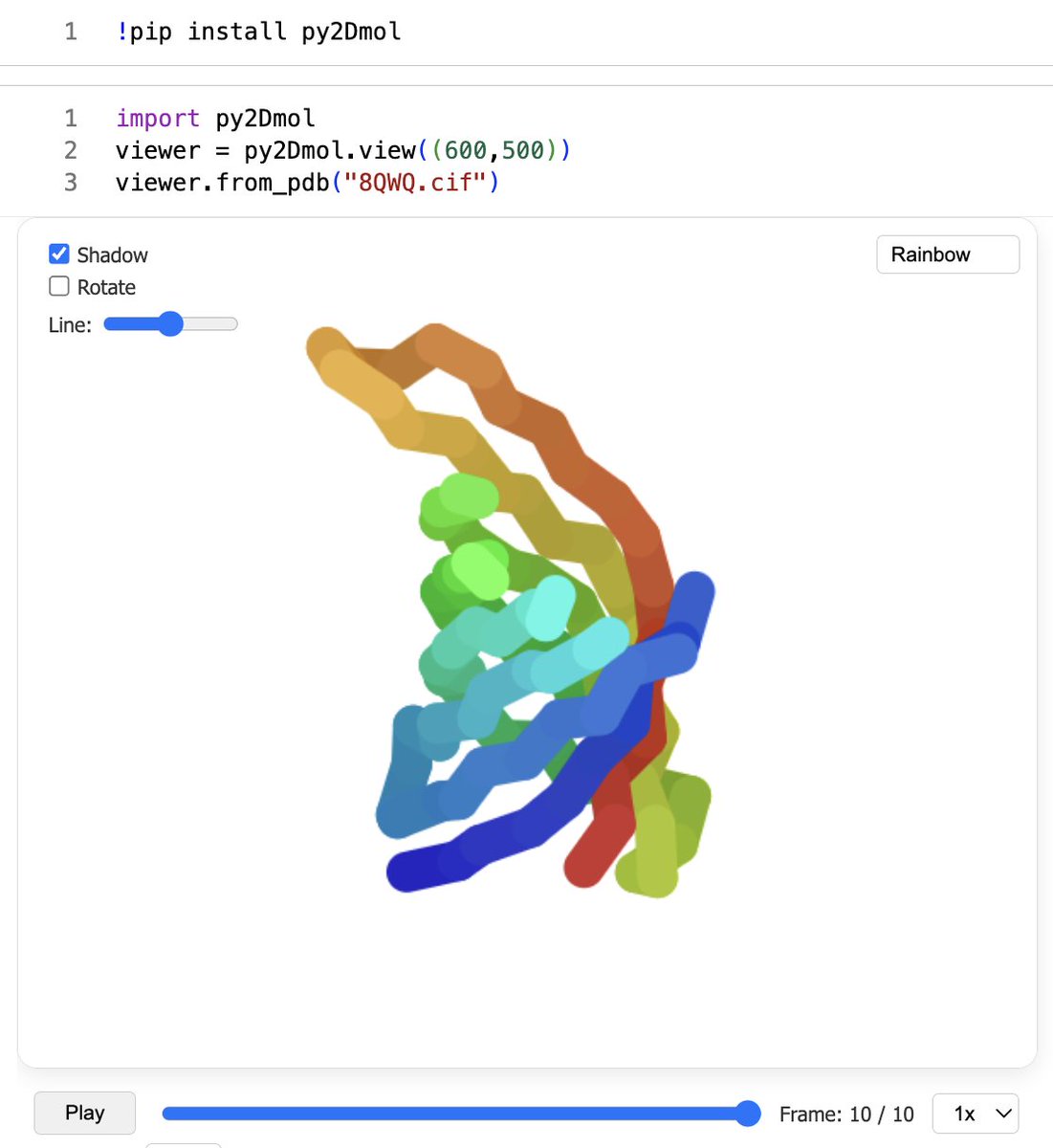
6
42
297
19,736
Jordan Hoff retweeted

28 Oct 2025
We're thrilled to announce SeqHub, an AI-enabled platform for biological sequence analysis. SeqHub brings together sequence search, genome annotation, and data sharing in one place.
I dreamed of a single place where I could learn everything about my sequences. Today, a much more refined version of this dream takes form with SeqHub.org, built by an incredible team at @tatta_bio. Our goal is to make sequence interpretation more intuitive and collaborative for everyone working with biological sequences.
Currently, SeqHub is optimized for microbial protein and genome analysis. As we expand beyond microbial data, we'd love your feedback to help shape what comes next. I'm deeply grateful to our team at Tatta Bio, and to our collaborators and funders, for making this vision a reality.
Check it out at seqhub.org!
14
114
558
53,010
Jordan Hoff retweeted

1 Oct 2025
Today in @Nature , we highlight how a cousin of CRISPR-Cas10, mCpol, establishes an evolutionary trap in anti-phage immune systems.
Check out Erin Doherty's and my work from Doudna lab here:
nature.com/articles/s41586-0…
3
39
132
10,227

Welcome to the age of generative genome design!
In 1977, Sanger et al. sequenced the first genome—of phage ΦX174.
Today, led by @samuelhking, we report the first AI-generated genomes. Using ΦX174 as a template, we made novel, high-fitness phages with genome language models. 🧵
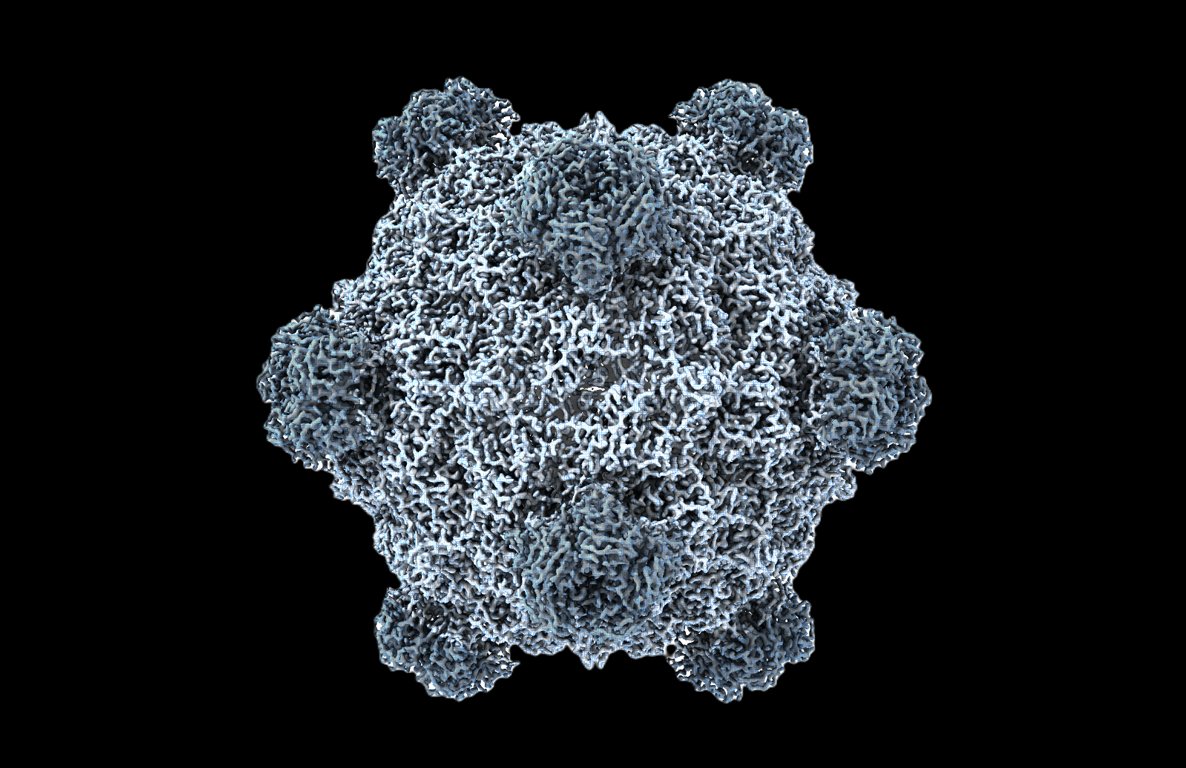
35
208
1,010
98,712
Jordan Hoff retweeted

5 Aug 2025
Excited to share work with @ZhidianZ, Milot Mirdita, Martin Steinegger, and @sokrypton
biorxiv.org/content/10.1101/…
TLDR: We introduce MSA Pairformer, a 111M parameter protein language model that challenges the scaling paradigm in self-supervised protein language modeling
🧵
8
52
194
26,394
Jordan Hoff retweeted
26 Apr 2025
Thanks Abhi @owl_posting for a wonderful conversation! It was a an honor to be the 3rd guest on the show. 😎
25 Apr 2025

What could Alphafold 4 look like? (Sergey Ovchinnikov, Ep #3)
2 hours listening time
(links below)
To those in the (machine-learning for protein design) space, Dr. Sergey Ovchinnikov (@sokrypton) is a very, very well-recognized name.
A recent MIT professor (circa early 2024), he has played a part in a staggering number of recent great papers in the field: ColabFold, RFDiffusion, Bindcraft, automated design of soluble proxies of membrane proteins, elucidating what protein language models are learning, conformational sampling via Alphafold2, and many more. Of course, all these papers were group efforts, but Sergey's name comes up astonishingly frequently!
And even beyond the research that have come from his lab in the last few years, the co-evolution work he did during his PhD/fellowship also laid some of the groundwork for the original Alphafold paper, being cited twice in it.
This is a two hour conversation with him, asking every question I could think of. We talk about his own journey into biology research, an issue he has with Alphafold3, what Alphafold4-and-beyond models may look like, what research he’d want to spend a hundred million dollars on, and lots more.
Topics/institutions we discuss: @arcinstitute's Evo models, @HWaymentSteele's work, @IsomorphicLabs's AF2/AF3, and @EvoscaleAI's ESM models
Also, extremely grateful to Asimov Press (@asimovpress) for helping fund the travel studio time required for this episode! They are a non-profit publisher dedicated to thoughtful writing on biology and metascience, such as articles over synthetic blood and interviews with plant geneticists. I myself have published within them twice! I highly recommend checking out their essays at asimov.press, or reaching out to editors@asimov.com if you’re interested in contributing.
Timestamps:
[00:00:00] Highlight clips
[00:01:10] Introduction Sergey's background and how he got into the field
[00:18:14] Is conservation all you need?
[00:23:26] Ambiguous vs non-ambiguous regions in proteins
[00:24:59] What will AlphaFold 4/5/6 look like?
[00:36:19] Diffusion vs. inversion for protein design
[00:44:52] A problem with Alphafold3
[00:53:41] MSA vs. single sequence models
[01:06:52] How Sergey picks research problems
[01:21:06] What are DNA models like Evo learning?
[01:29:11] The problem with train/test splits in biology
[01:49:07] What Sergey would do with $100 million
3
18
147
11,230