
I send you forth as sheep in the midst of wolves: be ye therefore wise as serpents, and harmless as doves.
Joined September 2024
- Tweets 786
- Following 170
- Followers 111
- Likes 1,452
40 Photos and videos










billwilliam retweeted
According to Fantini et al., ganglioside-binding motifs typically follow the pattern of triad alternating basic and aromatic residues such as (K,R)-Xn-(F,Y,W)-Xn-(K,R), in which the spacer Xn is usually composed of four to five residues. [1] They identified over-representation of aromatic and basic residues in residues 129-158 of SARS-CoV-2 spike. [1] The ganglioside-binding domain of SARS-CoV-2 spike is constituted by residues 111-162. [2] Aromatic sidechains form pi-pi stacking against sugar ring of ganglioside, while positively charged, basic sidechains interact with carboxylic acid moieties on terminal sialic acid.
The Pradhan insert 144-YYHKNNKS-151 probably makes SARS-CoV-2 spike a better ganglioside-binding motif, while SARS-CoV-1 spike lacks insert at this position. As shown by sequence alignment, MERS spike protein contains sequence (FYCILEPR) at the Pradhan position. The motif on MERS spike is also composed of alternating aromatic and basic residues, right?
It’s not surprising that MERS spike also binds ganglioside. Both SARS-CoV-2 and MERS spike proteins are characterized by over-presentation of basic and aromatic residues at the NTD ganglioside-binding domain.
References:
1. Fantini J, Di Scala C, Chahinian H, Yahi N. Structural and molecular modelling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int J Antimicrob Agents. 2020 May;55(5):105960. doi: 10.1016/j.ijantimicag.2020.105960. Epub 2020 Apr 3. PMID: 32251731; PMCID: PMC7128678.
pubmed.ncbi.nlm.nih.gov/3225…
2. Fantini J, Chahinian H, Yahi N. Leveraging coronavirus binding to gangliosides for innovative vaccine and therapeutic strategies against COVID-19. Biochem Biophys Res Commun. 2021 Jan 29;538:132-136. doi: 10.1016/j.bbrc.2020.10.015. Epub 2020 Oct 10. PMID: 33097184; PMCID: PMC7547605.
pubmed.ncbi.nlm.nih.gov/3309…

2
2
68
billwilliam retweeted

Thank you Kevin... 🙏🙏🙏
@GauteNilsen, all blood products (albumin, plasma, etc.) must be considered contaminated until we have systematic ultrasensitive testing for Spike and its amyloid fibrils.
Vaccinated and unvaccinated blood must be screened before any use. The problem is the SARS-CoV-2 Spike protein - whatever its origin. And the Spike protein does not necessarily float freely; it can bind and form stable complexes with the albumin present in blood donations.
pmc.ncbi.nlm.nih.gov/article…
2
10
16
353
billwilliam retweeted

Jun 12
China attacked our diplomats in Guangzhou late last decade with sonic weapons, causing injuries. Our presidents did nothing in response.
79
112
618
44,875
billwilliam retweeted

Jun 11
NEW docs declassified by Tulsi Gabbard and released by Rand Paul on Fauci, COVID, and the Biodefense Blob. 🧵
Why did the U.S. work with a military lab in Wuhan?
A CIA report sets the stage: Bioweapons are harder to detect than nuclear weapons. Virologists should collect intel through conferences and labs.


25
238
606
40,606
billwilliam retweeted

Jun 10
Director of National Intelligence Tulsi Gabbard in coming days is expected to release declassified intelligence documents on the origin of the COVID-19 virus -- a disclosure that comes after years of disputed and controversial assessments on whether the pandemic originated in a Chinese laboratory or came from a wild animal. trib.al/6gaO1ni
5
17
36
2,138
billwilliam retweeted

New email released --

"Tony, Jeremey, Larry & I [Collins] helped with" Proximal Origins but "are appropriately not mentioned"
🤣😂
@julianbarnes @SherylNYT @NewsfromScience @johnstravis @jeffmervis @sciencecohen @RetractionWatch @NatureMedicine @Nature @ScienceMagazine @ewinsberg @Marcia4Science

4
5
30
1,695
billwilliam retweeted

Jun 8
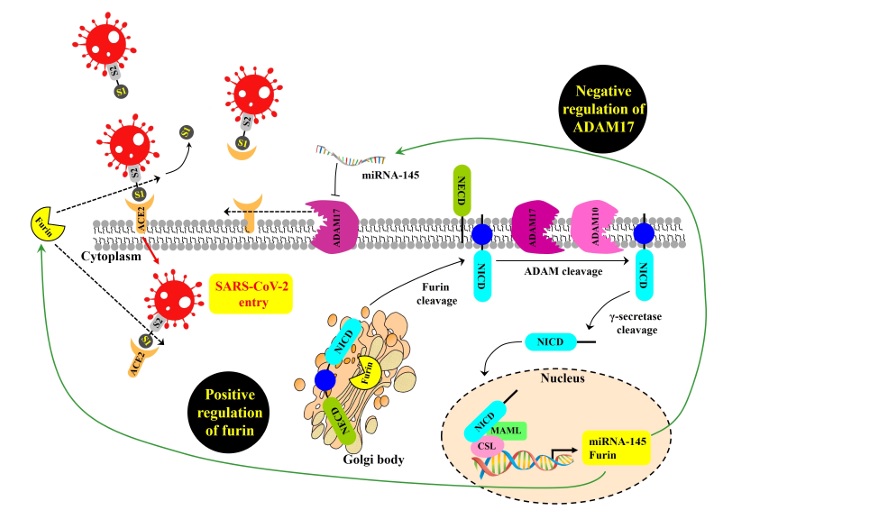
Notch signaling increases infection rate of SARS-CoV-2. After ligand binding and subsequent proteolysis by ADAM10/ADAM17, the liberated Notch intracellular domain (NICD) translocates into the nucleus to form transcriptional complex, which induces expression of genes including furin and miRNA-145. [1] In other words, viral activation of Notch upregulates furin and makes furin cleavage site even more efficient. Moreover, furin also cleaves immature Notch protein to mature herodimeric form for cell surface expression. [1] Notch signaling and furin form positive feedback loop.
In addition, miRNA145 inhibits ADAM17 expression level. ADAM17 mediates proteolytic cleavage and ectodomain shedding ACE2, and soluble ACE2 serves as decoy blocking viral spike protein. Activation of Notch signaling inhibits this innate antiviral mechanism. [1]
Hyperinflammation forms self-sustaining positive feedback loop. NOTCH1 can bind IL-6 promoter directly to activate its transcription, and IL-6 in turn triggers expression of NOTCH ligands such as DLL1 and DLL4 (Delta-like ligands). [1] TNF-α also upregulates expression of NOTCH1 and NOTCH4. [1]
Besides the harmful effects, Notch signaling is beneficial by aiding adaptive immune protection, immune memory generation, and antibody production. [1] Notch signaling is also helpful for tissue regeneration in the lungs after viral infection. Activation of Notch enhances proliferation of basal cells necessary for healing and regeneration of other lung cell types. [1] However, excessive Notch activation perturbs balance of cell differentiation and promotes formation of fibrosis and honeycomb-like cysts. [1]
Multiple viral factors may be involved in triggering Notch signaling. First of all, SARS-CoV-2 spike protein is known to trigger hyperinflammation by interaction with Toll-like receptors (especially TLR4). [2] TLR4 typically recognizes hydrophobic molecules such as bacterial lipopolysaccharide (LPS). Viral spike protein binding to host TLR4 produces sepsis-like inflammation, including production of IL-6 and TNF-α. This phenomenon may kick start the positive feedback loop between IL-6 and Notch.
Wang et al. found Porcine epidemic diarrhea virus (PEDV) activates Notch signaling (upregulation of mRNA levels of JAG-1, DLL4, Notch-1) primarily through ORF3. [3] ORF3 is a transmembrane ion channel that plays functions in viral replication and virulence. Perhaps ORF3 induced endoplasmic reticulum stress and subsequent Notch activation are a common feature among various coronaviruses. SARS-CoV-2 also contains ORF3a. Of note, some studies suggest SARS-CoV-2 ORF3a is a calcium ion channel, whereas SARS-CoV-1 homolog is a selective potassium channel. [4] SARS-CoV-2 ORF3a may be a more enhanced virulence factor that induces ER stress by allowing calcium ions to leak out.
Breikaa et al. suggest that 5’UTR of SARS-CoV-2 genome may interact with proteins associated with Notch2 receptor signaling. [5]
ORF7a and ORF8 are additional virulence factors that trigger hyperinflammation. They may enhance Notch signaling and positive feedback loop by upregulating IL-6. ORF8 mimics IL17a. [6]
Hypoxia from viral infection activates Notch. [5] Notch signaling also exacerbates SARS-CoV-2-associated blood coagulation. [5]
Notch activation is quite common among several different viruses such as Epstein-barr virus, influenza virus, RSV, HPV, Human T-cell leukemia virus type 1 (HTLV-1), Hepatitis C virus (HCV), HBV, HIV. [5]
Anyway, the conclusion is that Notch activation can be engineered artificially. Some mutations in the spike protein may enhance binding with TLR4. Given that the Wuhan lab and other labs have collected thousands of viral samples, they could have selected for high-virulence forms of ORF3 (high-efficiency calcium ion channel). Other virulence factors like ORF7a and ORF8 can be deliberately selected or engineered.
References:
1. Baindara P, Sarker MB, Earhart AP, Mandal SM, Schrum AG. NOTCH signaling in COVID-19: a central hub controlling genes, proteins, and cells that mediate SARS-CoV-2 entry, the inflammatory response, and lung regeneration. Front Cell Infect Microbiol. 2022 Aug 4;12:928704. doi: 10.3389/fcimb.2022.928704. PMID: 35992174; PMCID: PMC9386183.
pubmed.ncbi.nlm.nih.gov/3599…
2. Sahanic S, Hilbe R, Dünser C, Tymoszuk P, Löffler-Ragg J, Rieder D, Trajanoski Z, Krogsdam A, Demetz E, Yurchenko M, Fischer C, Schirmer M, Theurl M, Lener D, Hirsch J, Holfeld J, Gollmann-Tepeköylü C, Zinner CP, Tzankov A, Zhang SY, Casanova JL, Posch W, Wilflingseder D, Weiss G, Tancevski I. SARS-CoV-2 activates the TLR4/MyD88 pathway in human macrophages: A possible correlation with strong pro-inflammatory responses in severe COVID-19. Heliyon. 2023 Nov 17;9(11):e21893. doi: 10.1016/j.heliyon.2023.e21893. PMID: 38034686; PMCID: PMC10686889.
pubmed.ncbi.nlm.nih.gov/3803…
3. Wang Y, Yang S, Zhao Y, Tian S, Cao Q, Geng X, Yang M, Song X, Shang H, Liu S, Guo R, Li Y, Sun M, Hu M, Fan B, Li B. PEDV infection downregulates goblet cell differentiation through activating the Notch pathway. Vet Res. 2025 Aug 12;56(1):168. doi: 10.1186/s13567-025-01599-5. PMID: 40797223; PMCID: PMC12341102.
pubmed.ncbi.nlm.nih.gov/4079…
4. Zhang J, Ejikemeuwa A, Gerzanich V, Nasr M, Tang Q, Simard JM, Zhao RY. Understanding the Role of SARS-CoV-2 ORF3a in Viral Pathogenesis and COVID-19. Front Microbiol. 2022 Mar 9;13:854567. doi: 10.3389/fmicb.2022.854567. PMID: 35356515; PMCID: PMC8959714.
pubmed.ncbi.nlm.nih.gov/3535…
5. Breikaa RM, Lilly B. The Notch Pathway: A Link Between COVID-19 Pathophysiology and Its Cardiovascular Complications. Front Cardiovasc Med. 2021 May 26;8:681948. doi: 10.3389/fcvm.2021.681948. PMID: 34124207; PMCID: PMC8187573.
pubmed.ncbi.nlm.nih.gov/3412…
6. Wu X, Xia T, Shin WJ, Yu KM, Jung W, Herrmann A, Foo SS, Chen W, Zhang P, Lee JS, Poo H, Comhair SAA, Jehi L, Choi YK, Ensser A, Jung JU. Viral Mimicry of Interleukin-17A by SARS-CoV-2 ORF8. mBio. 2022 Apr 26;13(2):e0040222. doi: 10.1128/mbio.00402-22. Epub 2022 Mar 28. PMID: 35343786; PMCID: PMC9040823.
pubmed.ncbi.nlm.nih.gov/3534…


2
1
1
706
billwilliam retweeted
Jun 5
NEW: Tulsi Gabbard is actively working to declassify intelligence about the origins of COVID and Havana Syndrome by June 30, an ODNI official tells @DailyCaller. A CIA whistleblower recently testified that Fauci and Biden's intel chief suppressed evidence of a Wuhan lab leak.
19
119
456
32,801
billwilliam retweeted

On June 4, the world marks 37 years since the Chinese Communist Party ordered its troops to attack thousands of peaceful demonstrators in and around Tiananmen Square. Those who sacrificed to uphold their unalienable rights of free expression and peaceful assembly will be vindicated someday.
3,556
9,272
45,599
1,812,238
billwilliam retweeted

Jun 3
🚨 BREAKING: 🚨
Dr. Vincent Munster, a foreign born national virologist who works at the Rocky Mountain Laboratory under @NIH and Claude Kwe, one of his foreign born colleagues from Cameroon who also works at NIH in the same lab have officially been charged with smuggling monkeypox and other deadly pathogens (possibly Ebola) into the United States from the Democratic Republic of Congo and lying about it when confronted by Border Patrol!!!
@WhiteCoatWaste and I are totally vindicated after we broke this story several weeks ago about how NIH was covering up the fact that several foreign born virologists working at NIH under the Trump admin at Rocky Mountain Laboratory, a Biosafety Level 4 laboratory, snuck lethal pathogens into the US. They work at the same lab where an Ebola infected monkey bit an NIH researcher, and this was also covered up by @DrJBhattacharya and NIH.
According to the press release,
“On January 25, 2026, Munster and Kwe arrived at the McNamara Terminal at Detroit Metropolitan Airport with travel originating from Brazzaville, Republic of Congo, where an outbreak of monkeypox was occurring. Monkeypox is an infectious virus that can result in painful rash, enlarged lymph nodes, fevers and other ailments.
Munster and Kwe were inspected and interviewed by Customs and Border Protection (CBP) officials upon their arrival. CBP officers observed Kwe and Munster traveling with a large black plastic case. Munster and Kwe falsely told CBP officers that the black case contained diagnostics and testing equipment. But subsequent investigation by CBP and FBI agents revealed that the case actually contained 113 vials in Styrofoam coolers. As of the date of the complaint, the FBI has tested 20 of the 113 vials. Seventeen of them contained deactivated monkeypox virus, one contained the Chickenpox virus, and two contained only human DNA.
“These NIH experts apparently broke our laws by smuggling viral pathogens on a packed commercial airplane from an outbreak in the Republic of Congo. Let that sink in,” United States Attorney Gorgon stated.
“No researchers should believe their positions, credentials, or professional status place them above the law,” said Jennifer Runyan, Special Agent in Charge of the FBI Detroit Field Office. “The allegations in this case are serious. They involve the dangerous and unlawful smuggling of deactivated Mpox virus into the United States and alleged efforts to mislead our federal agents.”
The FBI admits only 20 of the 113 vials have been tested, which means we could soon learn that other lethal pathogens from Africa were illegally smuggled into our country by foreign born virologists who are Dr. Fauci acolytes.
Additionally, as I have previously reported, Munster’s wifeEmmie de Wit still works at NIH in the same laboratory where she studies Ebola. She should not be allowed to continue working at NIH, as White Coat Waste and I exposed she was also allegedly traveling with her husband and his colleague when they were caught at the airport in Michigan smuggling viruses into the United States from an outbreak zone in the Congo.
Munster and Kwe face a maximum sentence of five years in prison.
The investigation is being conducted by the Detroit Field Office of the Federal Bureau of Investigation, the U.S. Customs and Border Protection Detroit and the U.S. Department of Health and Human Services Office of Inspector General (HHS-OIG).
@TimSheehyMT @JustinRGoodman @SenRickScott
Read the full press release here:
justice.gov/usao-edmi/pr/fed…
May 24

NIH @NIH researcher Vincent Munster was recently caught at the airport in Michigan smuggling vials of deadly African pathogens, raising serious questions about whether his reckless operations sparked the current EBOLA outbreak.
White Coat Waste @WhiteCoatWaste Senior VP Justin Goodman @JustinRGoodman confirmed that Munster's rogue facility known as the Rocky Mountain Lab in Montana has a track record of severe containment failures, including a recent incident in which an Ebola-infected monkey bit a lab worker.
With gain-of-function lunatic and former Acting Director of NIAID Jeff Taubenberger now jumping ship to escape the fallout, HHS @HHSGov Secretary Robert F. Kennedy Jr. @RobertKennedyJr needs to PURGE the rest of Dr. Fauci's holdovers from HHS, NIH, and NIAID, and stop these mad scientists before more American lives are lost in yet another preventable scamdemic.
214
3,697
6,741
181,561
billwilliam retweeted
Love the abstract:
"SARS-CoV-2 has a mosaic genome, to which different progenitors contribute."
😂🤣 🔥🔥🔥🔥🔥🔥🔥🔥🔥🔥🔥
AKA a chimera!

4
6
20
1,812
billwilliam retweeted

This may be the reason for the PRRAR FCS.
Diabolical if so.
RRAR had already been shown to be very good at binding Neuropilin-1.
But not as good as RPAR, so why not use that?
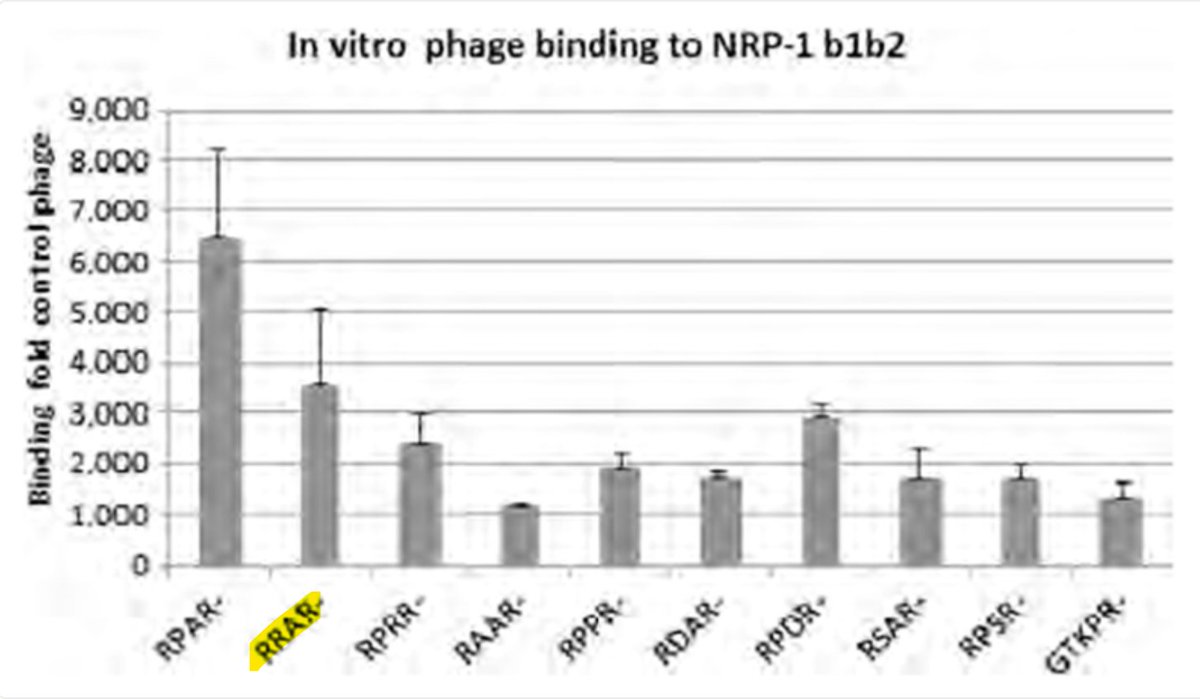
Mar 31

C end R Rule peptide binding studies, NRP-1, MHV central nervous system studies—researchers were so interested in Proline and RRAR




3
12
31
2,087
billwilliam retweeted
MERS MA30 was made by Kun Li, originally from Wuhan University, State Key Lab of Virology in Stanley Perlman's lab.

2
6
16
2,901
billwilliam retweeted

Excited to share our study by @keylas3 et al. on pathological autoantibodies in people with Long COVID. We asked whether IgG in patients with Long COVID bind to human tissues/antigens and cause pathologies when transferred into mice. With @PutrinoLab
doi.org/10.1016/j.cell.2026.…
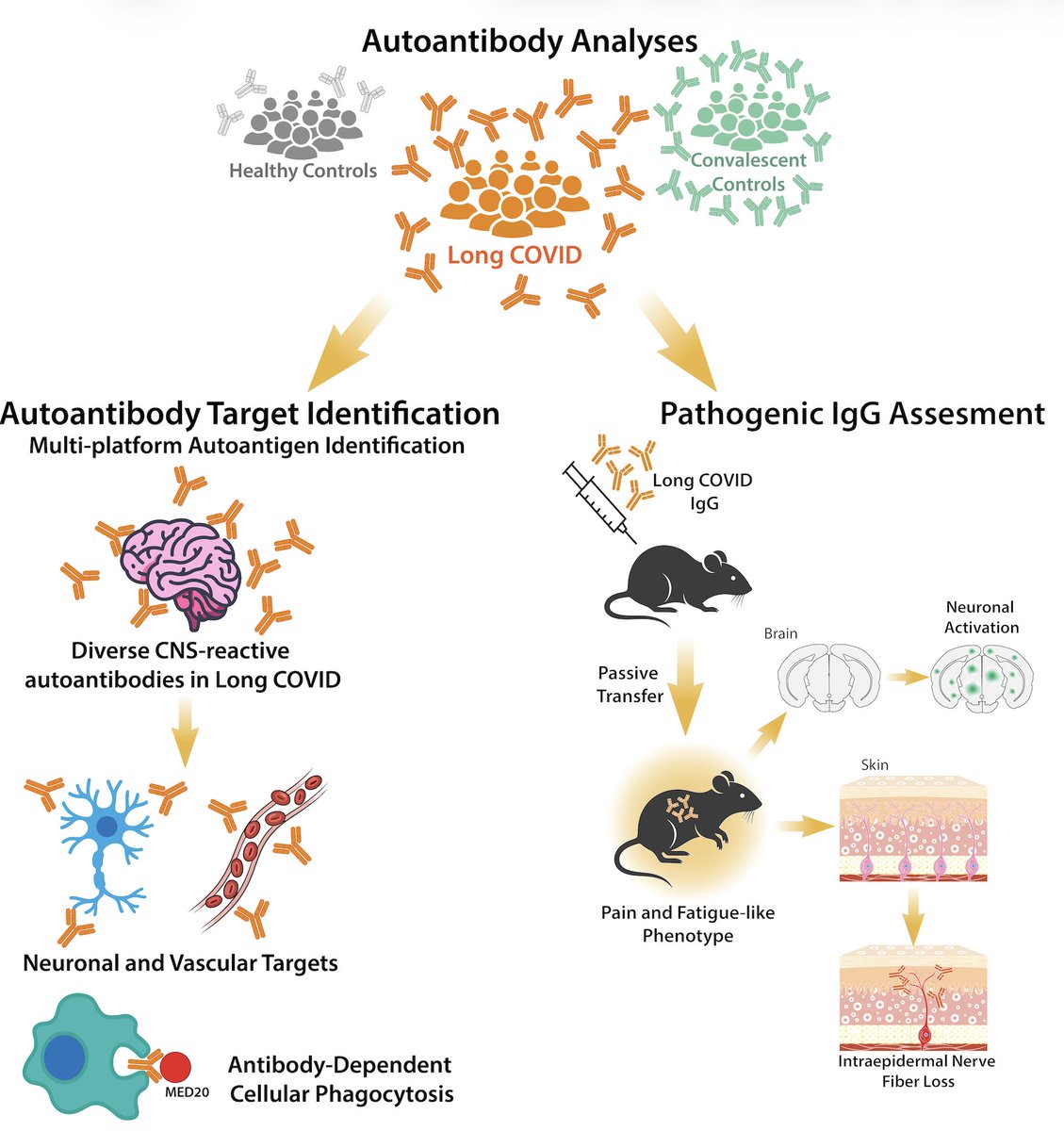
93
468
1,279
224,360
billwilliam retweeted
May 29
Mike previously pointed out that vanadium nanoparticles may inhibit SARS-CoV-2. The mechanism is based on electrostatic charge. Decavanadate (V10O28) is a polyanion that inhibits SARS-CoV-2 by interacting with positively charged aa on RBD or at furin cleavage site. [1] Despite promising result, vanadium nanoparticles still face the hurdle of heavy metal toxicity. The metal vanadium doesn’t matter—it’s the overall negative charge that mediates inhibition of positively charged patches on spike protein.
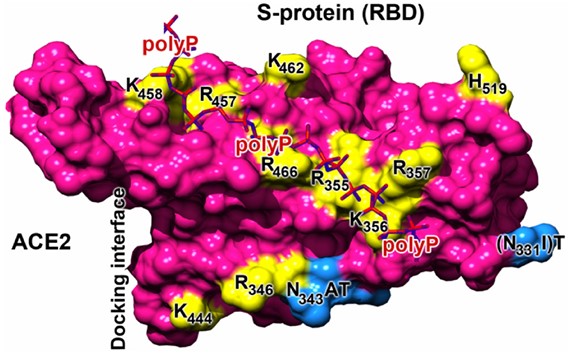
By the same token, polyphosphate is another anionic polymer that inhibits SARS-CoV-2 by interacting with a positively charged cleft at the RBD-ACE2 interface. [2-4] Polyphosphate is naturally produced in the human blood and in the platelets. Polyphosphate (polyP3 or polyP40) inhibits SARS-CoV-2 at physiological concentration. [2, 3] Even if polyphosphate may not be safe as injection, it should be safe as nasal rinse.
By the way, the ebola virus also contains furin cleavage site. I don’t know if polyphosphate can inhibit ebola. Some of the discoverers of polyphosphate antivirals are affiliated with Johannes Gutenberg University in Germany.
References:
1. Favre D, Harmon JF, Zhang A, Miller MS, Kaltashov IA. Decavanadate interactions with the elements of the SARS-CoV-2 spike protein highlight the potential role of electrostatics in disrupting the infectivity cycle. J Inorg Biochem. 2022 Sep;234:111899. doi: 10.1016/j.jinorgbio.2022.111899. Epub 2022 Jun 9. PMID: 35716549; PMCID: PMC9183239.
pubmed.ncbi.nlm.nih.gov/3571…
2. Müller WEG, Schröder HC, Neufurth M, Wang X. An unexpected biomaterial against SARS-CoV-2: Bio-polyphosphate blocks binding of the viral spike to the cell receptor. Mater Today (Kidlington). 2021 Dec;51:504-524. doi: 10.1016/j.mattod.2021.07.029. Epub 2021 Aug 2. PMID: 34366696; PMCID: PMC8326012.
pubmed.ncbi.nlm.nih.gov/3436…
3. Neufurth M, Wang X, Tolba E, Lieberwirth I, Wang S, Schröder HC, Müller WEG. The inorganic polymer, polyphosphate, blocks binding of SARS-CoV-2 spike protein to ACE2 receptor at physiological concentrations. Biochem Pharmacol. 2020 Dec;182:114215. doi: 10.1016/j.bcp.2020.114215. Epub 2020 Sep 6. PMID: 32905794; PMCID: PMC7474874.
pubmed.ncbi.nlm.nih.gov/3290…
4. Ferrucci V, Kong DY, Asadzadeh F, Marrone L, Boccia A, Siciliano R, Criscuolo G, Anastasio C, Quarantelli F, Comegna M, Pisano I, Passariello M, Iacobucci I, Monica RD, Izzo B, Cerino P, Fusco G, Viscardi M, Brandi S, Pierri BM, Borriello G, Tiberio C, Atripaldi L, Bianchi M, Paolella G, Capoluongo E, Castaldo G, Chiariotti L, Monti M, De Lorenzo C, Yun KS, Pascarella S, Cheong JH, Kim HY, Zollo M. Long-chain polyphosphates impair SARS-CoV-2 infection and replication. Sci Signal. 2021 Jul 6;14(690):eabe5040. doi: 10.1126/scisignal.abe5040. PMID: 34230209; PMCID: PMC8432949.
pubmed.ncbi.nlm.nih.gov/3423…
1
1
2
137
billwilliam retweeted
May 26
Ebola is already in the US…
Soon it will be announced.
May 26

Canada is no longer treating the Ebola outbreak as a distant regional event.
Enhanced airport screening is now active.
Quarantine officers have been deployed.
Travelers from affected regions are being flagged for assessment.
562
446
2,390
467,575
billwilliam retweeted
China's mobile BSL-3 was a rip-off of models they got from France after SARS.
Note the first two author on the paper are Yi-gang Tong and Weifeng Shi. They're the key authors of the pangolin covs and RmYN02 respectively. Although Weifeng Shi is a civilian based at Shandong University, he and Tong are long time collaborators. Director of the AMMS Beijing lab, Wuchun Cao is a Shandong graduate and guest professor.

1
2
2
85
billwilliam retweeted
May 25
@AnneliseBocquet @breakfast_dogs @SitSilver
A few pieces of circumstantial evidence:
1. In 2014, China deployed a mobile BSL-3 lab in Sierra Leone for Ebola virus diagnostics. The mobile lab—essentially a container carrying biotech equipment—can be mounted on truck or dismounted for operation on the ground, and is transportable by air, land, and sea. These biotech RVs, which carry onboard water tank, autoclave, and BSL-3 area, become operable when connected to water, gas, and electricity. [1] My first impression is that such containers may be military equipment (though I may be wrong). Some of the authors of the paper are affiliated with the Beijing Institute of Microbiology and Epidemiology—a PLA subsidiary.
Such mobile labs are OK if used for peaceful purposes, although the risk is that such containers may be abused to build illegal, underground labs.
2. In addition, China built a Sierra Leone-China Friendship Biological Safety Lab (permanent BSL-3 lab) for Ebola detection in Jui District, Freetown, Sierra Leone. [2] According to The Calabash Newspaper, “although the lab was originally intended to be handed over to Sierra Leone’s Ministry of Health and Sanitation after the Ebola crisis, financial and technical constraints led China CDC to retain its management.” [3] “Expert teams from China CDC have worked in annual rotations.” China CDC Country Director in Sierra Leone is Dr. Haimo Shen.
3. I guess some of China’s disease control team in Sierra Leone may be tied to the military. A 2021 paper on China CDC Weekly journal discussed the establishment of an electronic surveillance system for infectious diseases in Sierra Leone. Many of this paper’s authors are affiliated with the Beijing Institute of Microbiology and Epidemiology—a PLA subsidiary. This project was funded by the China Mega-Project on Infectious Disease Prevention (2018ZX10101003-002). [4]
References:
1. Zhang Y, Gong Y, Wang C, Liu W, Wang Z, Xia Z, Bu Z, Lu H, Sun Y, Zhang X, Cao Y, Yang F, Su H, Hu Y, Deng Y, Zhou B, Zhao Z, Fu Y, Kargbo D, Dafae F, Kargbo B, Kanu A, Liu L, Qian J, Guo Z. Rapid deployment of a mobile biosafety level-3 laboratory in Sierra Leone during the 2014 Ebola virus epidemic. PLoS Negl Trop Dis. 2017 May 15;11(5):e0005622. doi: 10.1371/journal.pntd.0005622. PMID: 28505171; PMCID: PMC5444861.
pmc.ncbi.nlm.nih.gov/article…
2. Wang Q, Zhou WM, Zhang Y, Wang HY, Du HJ, Nie K, Song JD, Xiao K, Lei WW, Guo JQ, Wei HJ, Cai K, Wang YH, Wu J, Kamara G, Kamara I, Wei Q, Liang MF, Wu GZ, Dong XP. Good laboratory practices guarantee biosafety in the Sierra Leone-China friendship biosafety laboratory. Infect Dis Poverty. 2016 Jun 23;5(1):62. doi: 10.1186/s40249-016-0154-5. PMID: 27333890; PMCID: PMC4918109.
pmc.ncbi.nlm.nih.gov/article…
3. “NPHA Awards Sierra Leone-China Friendship Lab for Outstanding Role in Mpox Response.” The Calabash Newspaper. October 24, 2025.
thecalabashnewspaper.com/nph…
4. Guangyu Zhao, Haorong Chen, Yanfeng Yan, Jiafu Jiang, Lei Lin, Baogui Jiang, Foday Sahr, Stephen Sevalie, Qiang Xu, Jinjin Chen, Henry Saidu Bangura, Kandeh Bassie Kargbo, Yajun Song, Wei Liu, Liqun Fang, Yansong Sun. The Establishment and Application of Mobile Electronic Surveillance System for Infectious Diseases with the Help of China — Sierra Leone, 2016-Present[J]. China CDC Weekly, 2021, 3(36): 763-768. DOI: 10.46234/ccdcw2021.189
weekly.chinacdc.cn/en/articl…


1
2
3
189
billwilliam retweeted

May 25
This new paper may be the most direct evidence so far that the SARS2 protein ORF3a can travel from the lungs to a distant organ through exosomes - in this case, the liver.
This is not a small detail. It may be one mechanism behind systemic COVID🧵

While SARS-CoV-2 primarily infects the respiratory tract, it can also damage other organs, including the liver, during both acute infection and long COVID1.
Liver involvement significantly increases the severity and mortality rate of COVID-19. nature.com/articles/s41421-0…
4
103
289
17,486