
PPIscreenML is a method for structure-based screening of protein-protein interactions using AlphaFold
1. Mischley et al. present PPIscreenML, a structure-based ML classifier that predicts whether a candidate protein pair truly interacts, using AlphaFold2-Multimer models as input rather than relying on sequence-only inference.
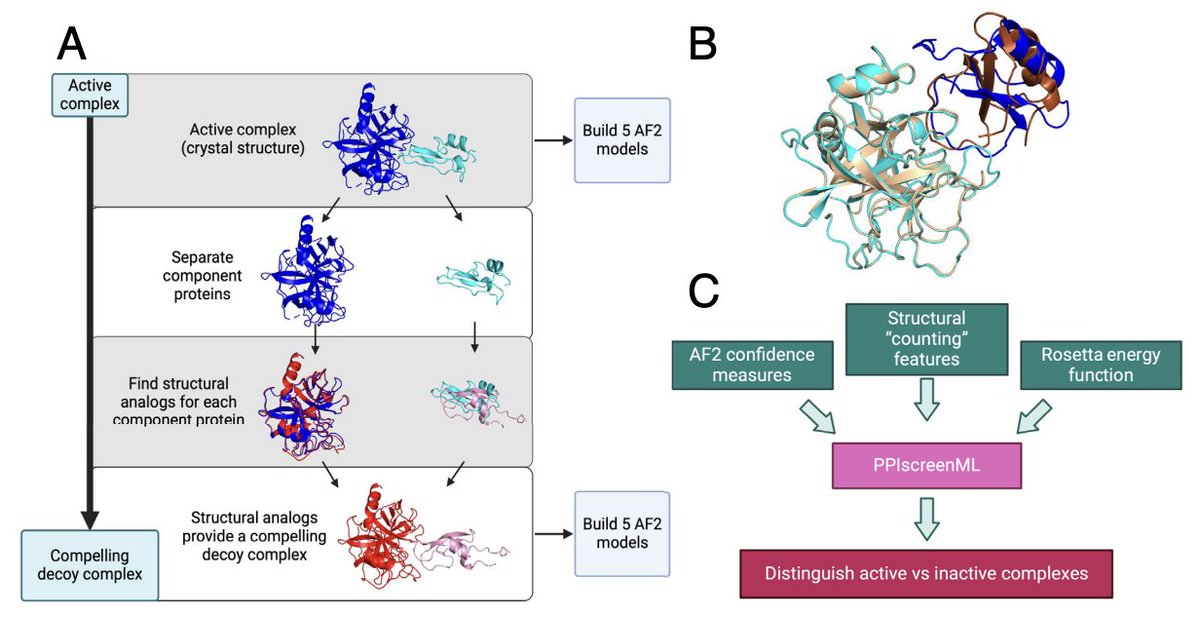
2. The key idea is to train explicitly on the interaction-vs-noninteraction task (not just “model quality”): PPIscreenML learns to separate AF2 models of real interacting heterodimers from AF2 models of “compelling decoys” that look structurally plausible.
3. Decoy generation is a central innovation: for each true PDB heterodimer, each partner is replaced by its closest structural analog (by TM-score) from a nonredundant set, then aligned into the original complex geometry—creating inactive pairs that mimic the geometry of true interfaces and are hard to dismiss by trivial heuristics.
4. Dataset scale and realism: 1481 nonredundant heterodimeric PDB complexes (<=30% sequence identity; excluding homodimers and antibody/antigen). Five AF2-Multimer v2.3 predictions per active and per decoy. Training includes only AF2 active models with DockQ >= 0.23 (to avoid learning from mis-docked “actives”), but the held-out test set keeps mis-docked actives to better reflect prospective screening.
5. Feature design blends AF2 confidence with physics-inspired energetics: 57 features total, spanning AF2 confidence metrics (pLDDT/pTM/PAE-derived terms), structural “counting” descriptors of interfaces, and Rosetta energy terms computed on the predicted complexes.
6. Model selection: several standard classifiers perform similarly, with gradient-boosted trees (XGBoost) best overall. On a completely held-out test set, the full feature model reaches ROC-AUC ~0.892 when scoring each candidate by the best of 5 AF2 models (a practical screening strategy).
7. A compact 7-feature version retains nearly the same performance (test ROC-AUC ~0.884), suggesting much of the signal is captured by a small set of interpretable interface cues: an interface-PAE statistic, interface charge count, and multiple Rosetta interfacial terms (LJ attractive/repulsive, solvation, electrostatics) plus a beta-sheet-related interface fraction.
8. Benchmark vs commonly used AF2-derived scores: on the same held-out test set, PPIscreenML outperforms iPTM and pDockQ for classifying interacting vs noninteracting pairs (AUC ~0.884 vs ~0.843 for iPTM and ~0.710 for pDockQ), highlighting the benefit of training specifically for screening rather than for structure-quality assessment.
9. Generalization test in a difficult “structurally conserved but selective” regime: across the TNF superfamily (18 ligands x 28 receptors = 504 pairs; only 36 known binders), AF2 can model many pairs in similar poses regardless of true binding. PPIscreenML nonetheless recapitulates specificity well (ROC-AUC ~0.93), with the top-scoring receptor matching a true interactor for 14/18 ligands (and within top-2 for 17/18), despite TNFSF not being in training (and training restricted to dimers).
💻Code: github.com/victoria-mischley…
📜Paper: doi.org/10.7554/eLife.98179
#ProteinProteinInteractions #AlphaFold #AlphaFoldMultimer #ComputationalBiology #StructuralBioinformatics #MachineLearning #Rosetta #Interactome #SystemsBiology #DrugDiscovery
1
2
25
11,363
PPIscreenML is a method for structure-based screening of protein-protein interactions using AlphaFold
1. Mischley et al. present PPIscreenML, a structure-based ML classifier that predicts whether a candidate protein pair truly interacts, using AlphaFold2-Multimer models as input rather than relying on sequence-only inference.
2. The key idea is to train explicitly on the interaction-vs-noninteraction task (not just “model quality”): PPIscreenML learns to separate AF2 models of real interacting heterodimers from AF2 models of “compelling decoys” that look structurally plausible.
3. Decoy generation is a central innovation: for each true PDB heterodimer, each partner is replaced by its closest structural analog (by TM-score) from a nonredundant set, then aligned into the original complex geometry—creating inactive pairs that mimic the geometry of true interfaces and are hard to dismiss by trivial heuristics.
4. Dataset scale and realism: 1481 nonredundant heterodimeric PDB complexes (<=30% sequence identity; excluding homodimers and antibody/antigen). Five AF2-Multimer v2.3 predictions per active and per decoy. Training includes only AF2 active models with DockQ >= 0.23 (to avoid learning from mis-docked “actives”), but the held-out test set keeps mis-docked actives to better reflect prospective screening.
5. Feature design blends AF2 confidence with physics-inspired energetics: 57 features total, spanning AF2 confidence metrics (pLDDT/pTM/PAE-derived terms), structural “counting” descriptors of interfaces, and Rosetta energy terms computed on the predicted complexes.
6. Model selection: several standard classifiers perform similarly, with gradient-boosted trees (XGBoost) best overall. On a completely held-out test set, the full feature model reaches ROC-AUC ~0.892 when scoring each candidate by the best of 5 AF2 models (a practical screening strategy).
7. A compact 7-feature version retains nearly the same performance (test ROC-AUC ~0.884), suggesting much of the signal is captured by a small set of interpretable interface cues: an interface-PAE statistic, interface charge count, and multiple Rosetta interfacial terms (LJ attractive/repulsive, solvation, electrostatics) plus a beta-sheet-related interface fraction.
8. Benchmark vs commonly used AF2-derived scores: on the same held-out test set, PPIscreenML outperforms iPTM and pDockQ for classifying interacting vs noninteracting pairs (AUC ~0.884 vs ~0.843 for iPTM and ~0.710 for pDockQ), highlighting the benefit of training specifically for screening rather than for structure-quality assessment.
9. Generalization test in a difficult “structurally conserved but selective” regime: across the TNF superfamily (18 ligands x 28 receptors = 504 pairs; only 36 known binders), AF2 can model many pairs in similar poses regardless of true binding. PPIscreenML nonetheless recapitulates specificity well (ROC-AUC ~0.93), with the top-scoring receptor matching a true interactor for 14/18 ligands (and within top-2 for 17/18), despite TNFSF not being in training (and training restricted to dimers).
💻Code: github.com/victoria-mischley…
📜Paper: doi.org/10.7554/eLife.98179
#ProteinProteinInteractions #AlphaFold #AlphaFoldMultimer #ComputationalBiology #StructuralBioinformatics #MachineLearning #Rosetta #Interactome #SystemsBiology #DrugDiscovery
13
913

Apr 12
Dissecting the Black Box of AlphaFold in Protein–Protein Complex Assembly
biorxiv.org/content/10.64898…
AlphaFold achieves high accuracy in predicting protein–protein complexes, yet the principles of their assembly remain unclear. Here, we present a unified interpretability framework for AlphaFold-Multimer and AlphaFold3 to dissect these mechanisms. We find that inter-protein coevolution is not a major driver; instead, complex formation is largely governed by monomer geometry and interface-level pattern matching, including backbone complementarity and residue interactions. Tracking the propagation of distance constraints during inference reveals a hierarchical process where monomer structures form first, followed by inter-chain interactions. This shows that cross-chain geometry is inferred from monomer features rather than coevolutionary signals. In antigen–antibody complexes, lower accuracy arises from flexible, noncanonical interfaces, highlighting conformational variability and atypical interactions as key challenges for improving immune complex prediction.
#AlphaFold3 #BioAI #AlphaFoldMultimer #MSA #AntibodyEngineering #StructuralBiology
1
5
442
Dissecting the Black Box of AlphaFold in Protein–Protein Complex Assembly
1. Li, Mu, and Yan present evidence that inter-protein coevolution (the usual explanation for AlphaFold complex success) is not the dominant driver of complex assembly in AlphaFold-Multimer or AlphaFold3; instead, assembly largely follows from monomer geometry plus interface-level matching.
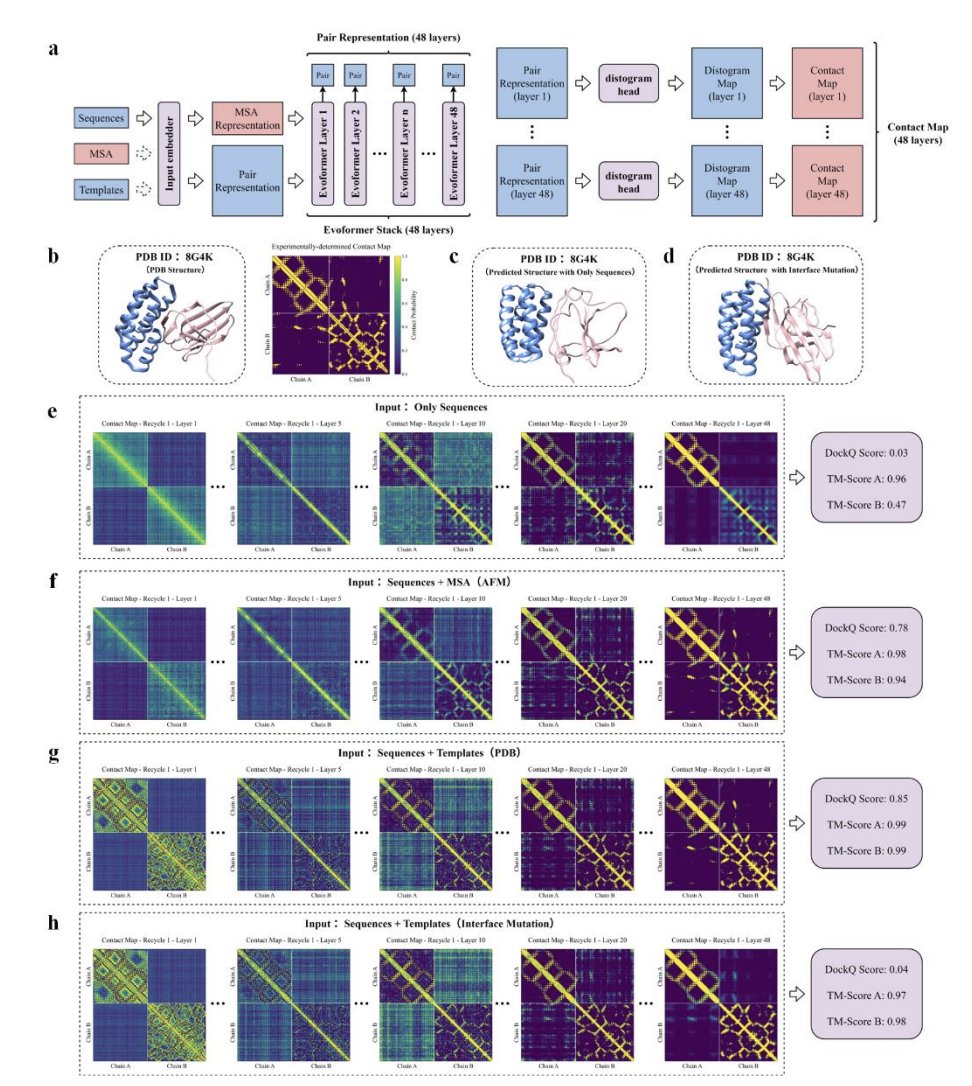
2. A time-segregated benchmark (PDB 2022-01-01 to 2024-12-20; training cutoff 2021-09-30) is built to reduce leakage: 200 homodimers and 316 heterodimers, evaluated mainly with DockQ across AFM and AF3.
3. Controlled MSA experiments separate “pairing” from “having MSAs”: AFM Paired MSA vs Block MSA (no cross-chain pairing) vs Randomly Paired MSA. Mean DockQ changes are minimal across these conditions, implying explicit paired-MSA coevolution contributes little for most targets.
4. The paper further removes potential “latent” inter-protein coevolution in unpaired MSAs by regenerating UniRef100 MSAs with species annotations and enforcing zero species overlap between partner MSAs; AFM/AF3 performance remains essentially unchanged, arguing against hidden species-level coevolution being a key signal.
5. The proposed mechanism: AlphaFold first establishes strong intra-chain geometric constraints (monomer folding/geometry), then infers inter-chain constraints downstream via geometric compatibility and interface sequence pattern matching; cross-chain organization is progressively refined through layers and recycling.
6. Template-driven tests support the geometry-first view: supplying high-quality bound-state monomer templates enables complex prediction accuracy comparable to MSA-based runs, and experimentally determined bound monomer templates perform even better; adding MSAs on top of such templates yields little additional gain.
7. A key nuance is “bound vs unbound” monomer geometry: predicted unbound monomer templates degrade complex accuracy, and the difference is concentrated at interface regions. Interface TM-score correlates with complex DockQ (reported Pearson r ≈ 0.575), highlighting interface conformation as a main determinant.
8. Interface residue identity is essential, not just backbone shape: mutating up to 10% of residues to glycine shows that interface mutations nearly abolish prediction accuracy under both MSA-based and template-based settings, while non-interface mutations have only moderate effects—consistent with a backbone sidechain “pattern matching” interface recognition.
9. The paper introduces AlphaFold-Constraint Propagation Mapping (AF-CPM), using OpenFold to extract Evoformer-layer pair representations and convert them (via the distogram head) into layer-wise contact probability maps (<12 Å). These visualizations show intra-chain constraints forming before inter-chain contacts, directly supporting hierarchical constraint formation.
10. For antigen–antibody complexes (154 nonredundant cases), paired MSAs still do not help; bound-state monomer templates help most. The limiting factor is attributed to immune-interface plasticity and atypical interface statistics (e.g., enrichment of Tyr/Trp on the antibody side), with CDR-H3 local accuracy strongly linked to docking success; AF-CPM suggests antigen–antibody assembly may require more recycling to converge as interface constraints emerge late.
💻Code: github.com/ChengfeiYan/AF-CP…
📜Paper: biorxiv.org/content/10.64898…
#AlphaFold #AlphaFoldMultimer #AlphaFold3 #ProteinComplexes #ProteinStructure #MSA #Interpretability #AntibodyEngineering #ComputationalBiology #StructuralBiology
12
58
4,182

13 Sep 2024
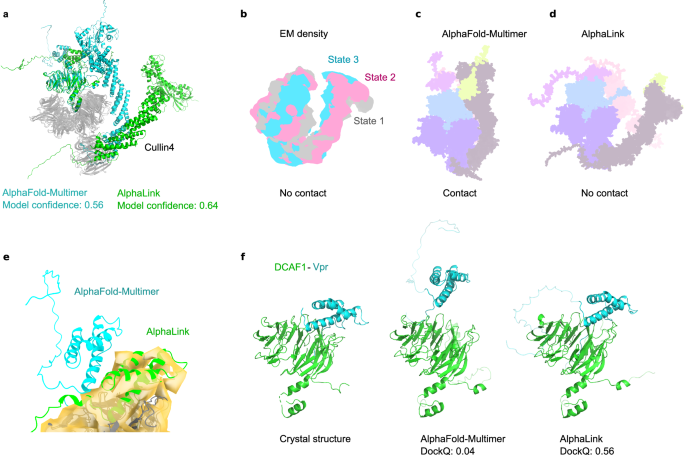
Authors @RappsilberLab @JorgStulke @rbolab and colleagues improve modelling performance on challenging targets by integrating experimental distance restraints from crosslinking mass spectrometry into AlphaFoldMultimer
nature.com/articles/s41467-0…
6
19
5,567

30 May 2023
@OPIGlets @brennanaba report that ImmuneBuilder is a set of deep learning models trained to predict the structure of antibodies, nanobodies, and T-Cell receptors with state-of-the-art accuracy while being much faster than AlphaFold2 and AlphaFoldMultimer. nature.com/articles/s42003-0…
1
3
116

20 Apr 2023
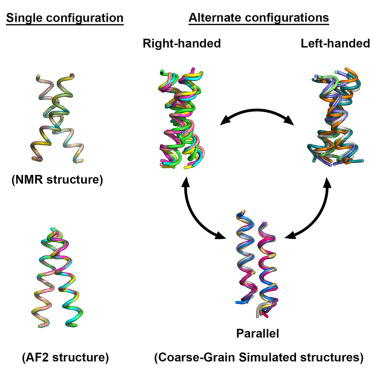
Nice to finally have this work out! Another nice collaboration with Matthias Buck and Amita Sahoo. published at @Structure_CP. Take a look here:
cell.com/structure/fulltext/…
#CoarseGrained #Martini3 #MDsimulations #AlphaFoldMultimer #TM_Peptides
1
3
27
3,610

19 Jan 2022
🚨New tool alert: AlphaFold Multimer🚨 We now offer the full #AlphaFold2 and #AlphaFoldMultimer on the COSMIC2 gateway. If you were having trouble predicting multimers on ColabFold, the larger GPUs on @SDSC_UCSD Expanse might be able to help! cosmic-cryoem.org/tools/alph…
2
33
137

7 Oct 2021
Another exciting #AI advancement in structural biology. #alphafold2 has now been adapted to predict multi-chain protein complexes. Whilst there still is room for improvement #alphafoldmultimer was able successfully predict the interface in 67% of cases.
deepmind.com/research/public…
1
2