
May 8
Excited to share that our paper “CodonMoE: DNA Language Models for Codon-Dependent mRNA Prediction” has recently been accepted by Bioinformatics! 🎉
CodonMoE is a lightweight plug-and-play adapter that enables existing DNA language models to perform mRNA analysis tasks effectively — without RNA-specific pretraining. Across tasks spanning mRNA stability, expression, and regulation, CodonMoE consistently improves DNA models while using substantially fewer parameters than specialized RNA models.🧬
📎 Paper: arxiv.org/abs/2508.04739
Big thanks to my collaborators and advisor: Litian Liang, Jiayi Li, and Carl Kingsford🙌
Comments and feedback are very welcome!
#GenomicLanguageModels #AIforScience #PEFT #mRNA
4
1
9
936
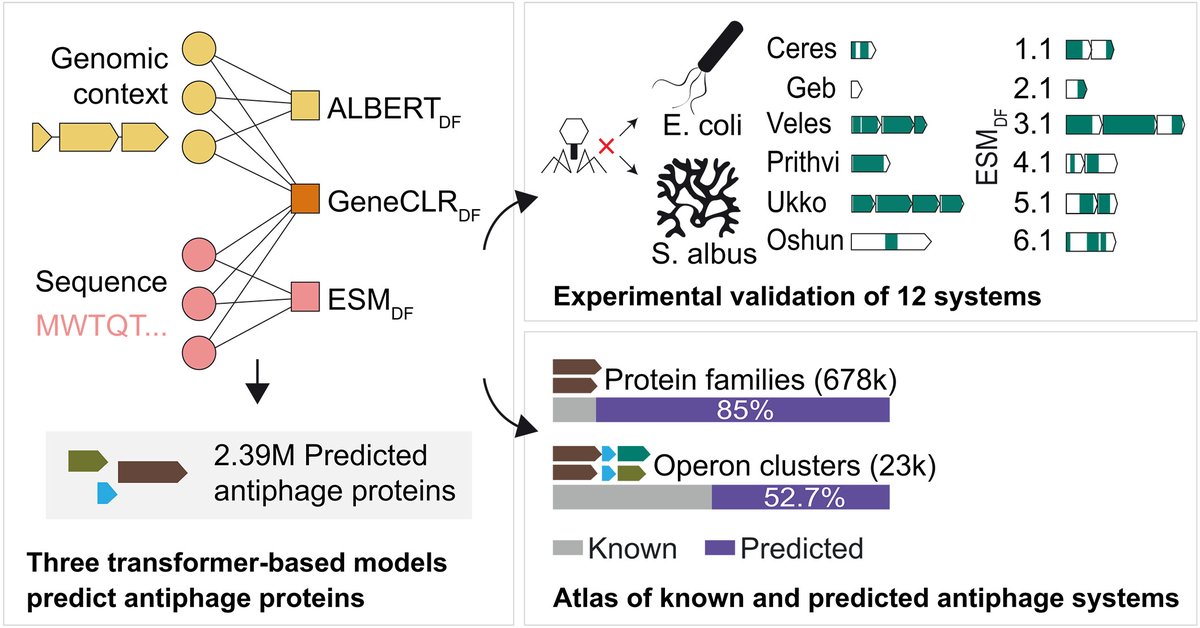
Protein and genomic language models uncover the unexplored diversity of bacterial immunity @ScienceMagazine
1. This study introduces three complementary deep-learning models—ALBERTDF, ESMDF, and GeneCLRDF—that predict antiphage defense systems by leveraging genomic context, protein sequence, or both, achieving up to 99% precision and 92% recall on curated benchmarks.
2. The hybrid model GeneCLRDF integrates protein sequence information with genomic neighborhood context through contrastive learning, outperforming both context-only and sequence-only approaches and enabling systematic discovery across the entire bacterial pangenome.
3. The researchers experimentally validated 12 newly predicted antiphage systems in Escherichia coli and Streptomyces albus, demonstrating that the framework recovers genuine defense mechanisms across phylogenetically distant bacteria, including systems with no prior association with immunity.
4. Applied to over 32,000 bacterial genomes, the models predict 2.39 million antiphage proteins and approximately 23,000 operon families, with more than 85% of predicted protein families having no previous link to antiviral defense.
5. The study reveals that approximately 1.5% of genes in a typical bacterial genome are devoted to antiviral defense—triple previous estimates—and confirms mobile genetic elements as major reservoirs of defensive systems.
6. The researchers provide an interactive, searchable atlas of predicted antiphage systems to enable community exploration and experimental follow-up, fundamentally expanding the known landscape of bacterial immunity.
💻Code: github.com/mdmparis/defense-…
📜Paper: science.org/doi/10.1126/scie…
#BacterialImmunity #PhageDefense #MachineLearning #Bioinformatics #GenomicLanguageModels #ProteinLanguageModels #AntiphageSystems #ComputationalBiology
4
28
2,175
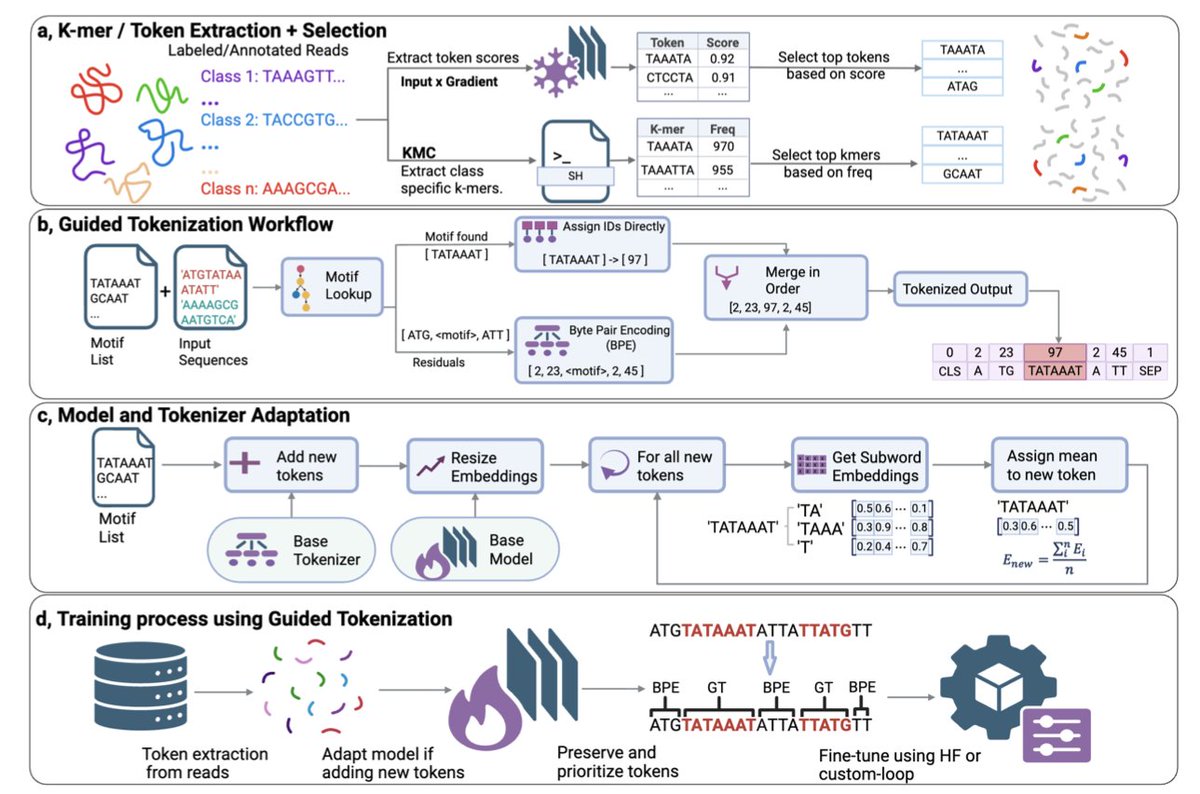
Guided Tokenization and Domain Knowledge Enhance Genomic Language Models' Performance
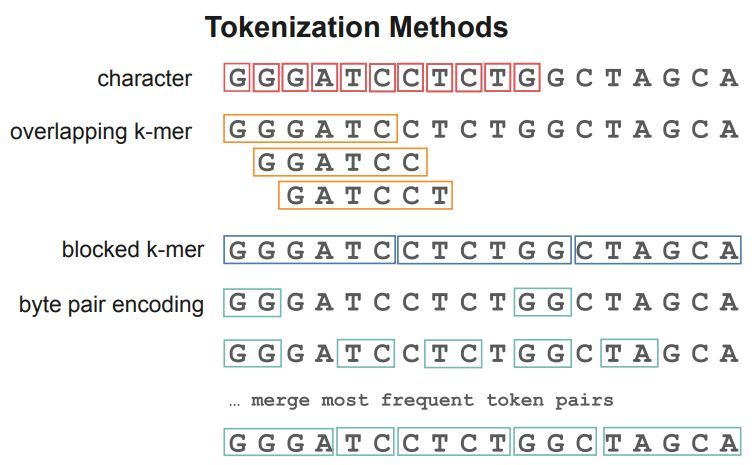
1. The authors introduce Guided Tokenization (GT), a novel domain-aware tokenization strategy that prioritizes biologically meaningful subsequences over standard fixed-length k-mers or Byte Pair Encoding, addressing a critical limitation where conventional methods fragment functionally important motifs like the TATA box in promoter sequences.
2. GT operates through a three-phase pipeline: extracting important tokens via gradient attribution or class-specific k-mer analysis, augmenting the tokenizer and model embeddings with mean subword initialization, and implementing a trie-based motif preservation algorithm that achieves O(n) time complexity for efficient sequence processing.
3. The approach demonstrates substantial performance gains across diverse genomic tasks, achieving 82.88% F1-score versus 78.93% for standard BPE in promoter detection, and 94.48% accuracy in multi-class antibiotic resistance gene classification compared to 92.28% for BPE, while substantially outperforming established tools like ResFinder and DeepARG.
4. For 16S rRNA taxonomic classification involving 4,288 genera, the authors developed a hierarchical ensemble approach combining order-level and genus-level classifiers, enabling GT to achieve 93.47% accuracy and demonstrating scalability strategies for high-dimensional classification spaces.
5. The study reveals that GT's effectiveness is modulated by the ratio of biological classes to vocabulary capacity, performing optimally when 10-30% vocabulary expansion accommodates class-specific k-mers, with particular advantages in data-scarce scenarios where domain-specific motifs compensate for limited training examples.
6. The methodology includes careful embedding initialization using mean-pooled subword representations rather than random initialization, enabling more effective transfer of pretrained knowledge and faster convergence during fine-tuning of compact genomic language models like DNABERT2-117M and seqLens-87M.
7. Comprehensive evaluation across binary and multi-class classification tasks demonstrates that GT not only improves accuracy but also enhances model calibration, with lower Brier scores indicating more reliable probability estimates for downstream genomic applications.
💻Code: github.com/omicsEye/guided_t…
📜Paper: biorxiv.org/content/10.64898…
#GenomicLanguageModels #Bioinformatics #Tokenization #DeepLearning #ComputationalBiology #Metagenomics #AntibioticResistance #16SrRNA #PromoterDetection #MachineLearning
2
12
1,011

11 Dec 2025
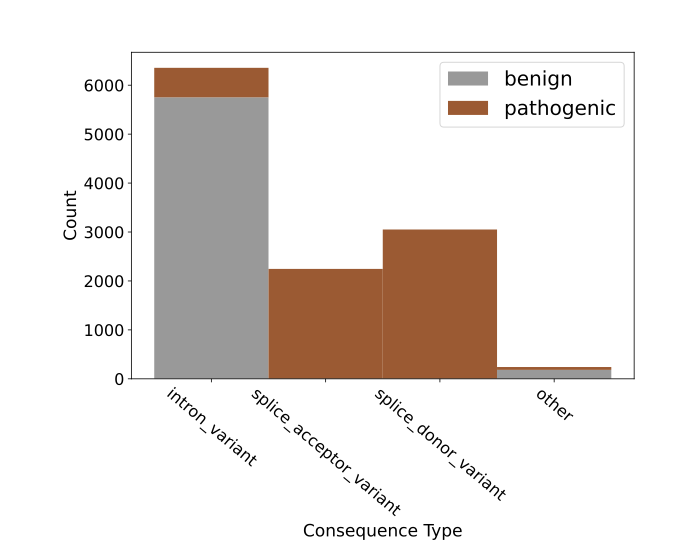
GUANinE v1.1 Reveals Complementarity of Supervised and Genomic Language Models
1. The GUANinE v1.1 benchmark provides a comprehensive evaluation of supervised and unsupervised genomic models, revealing that each excels in different tasks. Supervised models dominate functional annotation tasks like chromatin accessibility, while unsupervised models perform better in evolutionary conservation tasks.
2. The study introduces two new large-scale variant interpretation tasks: cadd-snv for measuring proxy deleteriousness and clinvar-snv for clinical pathogenicity. Conservation scores and language models dominate deleteriousness prediction, but translating this to pathogenicity remains challenging.
3. The research highlights that input context size and model parameter count trade off when compute budget is fixed. Models with larger context sizes don't always outperform those with fewer parameters, suggesting that parameter density and model complexity are crucial factors.
4. GUANinE v1.1 demonstrates that training on distal evolutionary sequences can improve model performance, even for human-specific tasks. This suggests that leveraging diverse genomic data can enhance the robustness of genomic language models.
5. The study also explores the potential of combining supervised and unsupervised approaches, showing that hybrid models may define the next era of genomic sequence modeling. An ensemble of NT-v2-500m and Sei outperformed either model alone on variant effect prediction tasks.
📜Paper: biorxiv.org/content/10.64898…
#Genomics #MachineLearning #Benchmarking #GenomicLanguageModels #SupervisedLearning #UnsupervisedLearning
1
2
3
790
7 Nov 2025
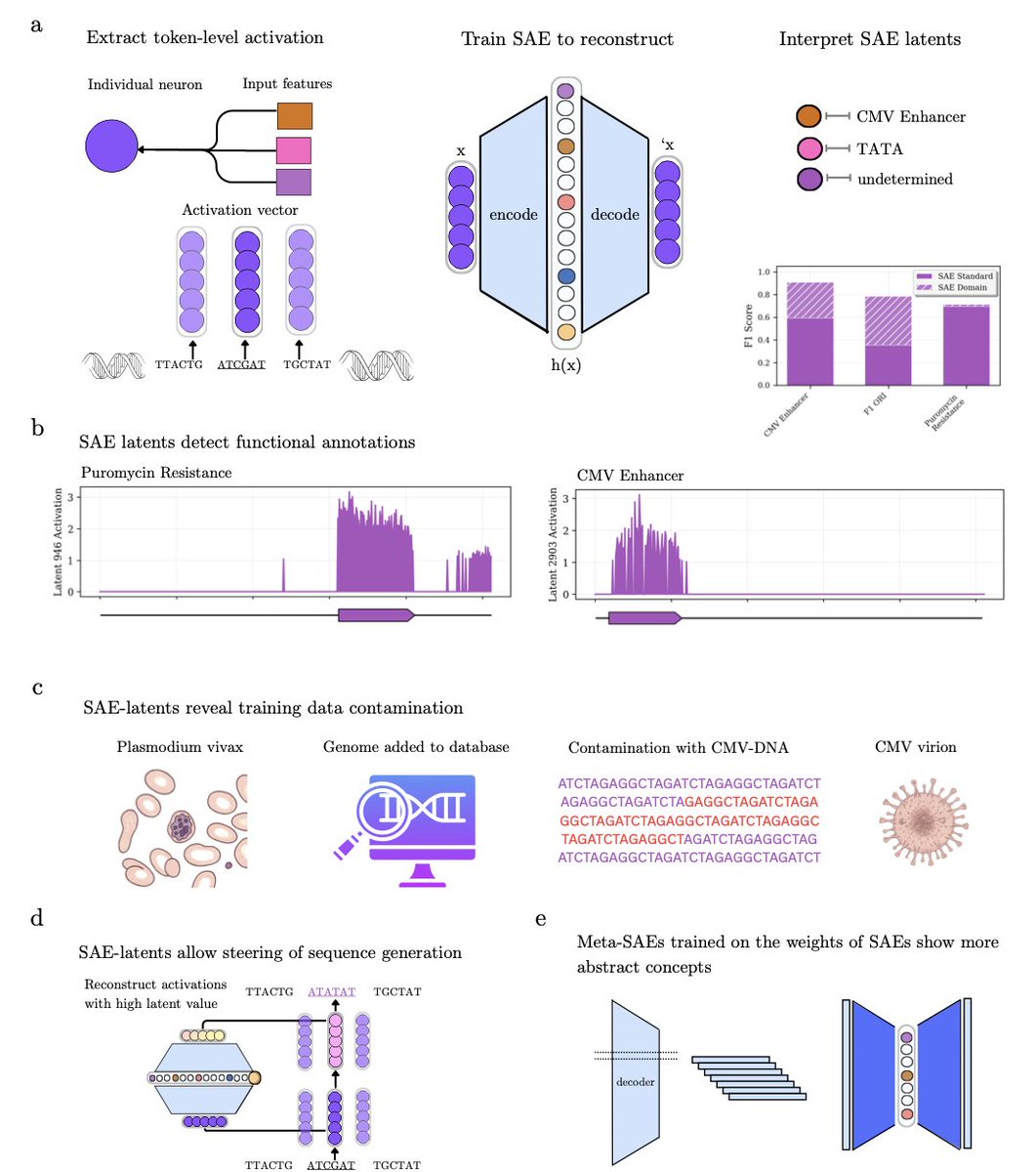
Decode-gLM: Tools to Interpret, Audit, and Steer Genomic Language Models
1. A new study introduces sparse autoencoders (SAEs) as a powerful tool for interpreting genomic language models. SAEs can decompose complex model activations into interpretable biological features without supervision, revealing over 100 diverse functional annotations encoded within the model.
2. The study demonstrates that SAEs can uncover hidden data leakage in training datasets. For example, viral regulatory elements like the CMV enhancer were detected in the model despite viral genomes being excluded from training, highlighting potential contamination in reference databases.
3. Meta-SAEs, trained on the decoder weights of SAEs, identify conceptual hierarchies within the model. This includes abstract features related to multiple HIV annotations, showcasing the model’s ability to encode coordinated biological processes.
4. The research also shows that SAEs can steer model predictions in biologically meaningful ways. For instance, an antibiotic-resistance feature was used to direct the model toward a specific aminoglycoside-resistance mutation in the 16S rRNA gene.
5. The findings establish SAEs as a versatile method for enhancing the interpretability and trustworthiness of genomic models. The authors provide a web interface for readers to explore the discovered features, promoting transparency and further research in this domain.
📜Paper: biorxiv.org/content/10.1101/…
#Genomics #AI #Interpretability #Bioinformatics #GenomicLanguageModels
5
25
2,420
11 Sep 2025
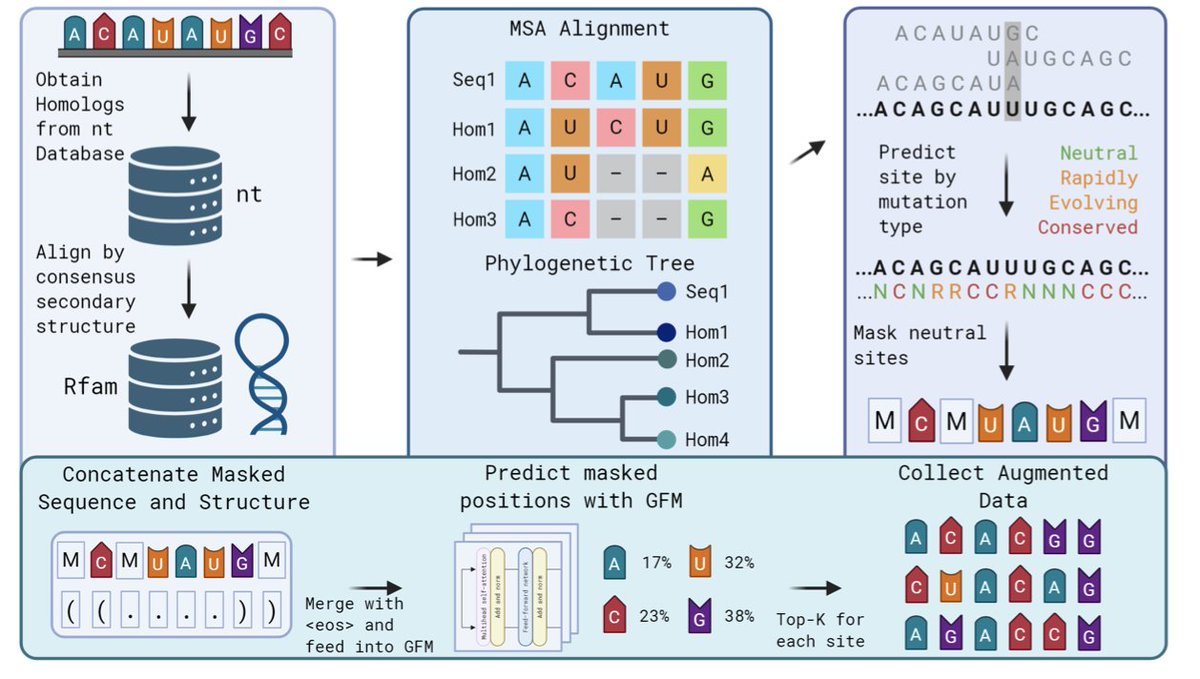
PhyloAug: An Evolutionary and Structure-Aware Data Augmentation Tool
1. The scarcity of high-quality genomic data is a significant challenge for Genomic Language Models (GLMs). PhyloAug addresses this by combining evolutionary signals with RNA secondary structure to generate biologically plausible augmentations that retain original function and structural coherence.
2. PhyloAug leverages the neutral theory of molecular evolution, identifying sites where mutations are unlikely to alter function. It integrates phylogenetic analysis via PAML to provide site-wise restrictions based on evolutionary principles, ensuring that augmentations do not disrupt critical functional motifs.
3. The method incorporates RNA secondary structure predictions to preserve RNA-protein interactions and prevent the model from learning impeding sequences. This is crucial for maintaining the biological relevance of the augmented data.
4. Experiments show that PhyloAug significantly improves the performance of GLMs on downstream tasks, with improvements up to 12.9% MCC. It also demonstrates a high level of sequence similarity to the underlying multiple sequence alignments, validating its biological validity.
5. PhyloAug is a versatile tool that can be applied to any Genomic Foundation Model through fine-tuning. It provides a one-size-fits-all solution for non-coding RNA tasks, enhancing model performance and robustness across various RNA-based tasks.
📜Paper: biorxiv.org/content/10.1101/…
#PhyloAug #GenomicLanguageModels #DataAugmentation #EvolutionaryBiology #RNASecondaryStructure #Bioinformatics
1
2
16
1,404

31 Aug 2025
The Impact of Tokenizer Selection in Genomic Language Models. #GenomicLanguageModels #Genomics #LLMs #Bioinformatics
academic.oup.com/bioinformat…
1
32
143
8,823
9 Aug 2025
CodonMoE: DNA Language Models for mRNA Analyses
1. CodonMoE introduces a novel lightweight adapter that transforms DNA language models into effective RNA analyzers without RNA-specific pretraining. This approach bridges the gap between DNA and RNA modeling, leveraging abundant DNA data for RNA tasks.
2. Theoretical analysis establishes CodonMoE as a universal approximator at the codon level, capable of mapping arbitrary functions from codon sequences to RNA properties given sufficient expert capacity.
3. Across four RNA prediction tasks spanning stability, expression, and regulation, DNA models augmented with CodonMoE significantly outperform their unmodified counterparts, achieving state-of-the-art results with 80% fewer parameters than specialized RNA models.
4. The CodonMoE architecture maintains sub-quadratic complexity while achieving superior performance. It integrates seamlessly with existing DNA models, making it a versatile plug-and-play module for mRNA analysis.
5. This work provides a principled path toward unifying genomic language modeling, reducing computational overhead while preserving modality-specific performance. It demonstrates the potential of DNA models to handle RNA tasks effectively with minimal adaptation.
📜Paper: arxiv.org/abs/2508.04739v1
#GenomicLanguageModels #RNAAnalysis #DNAData #ComputationalBiology #Bioinformatics

7
15
1,546
15 Jul 2025
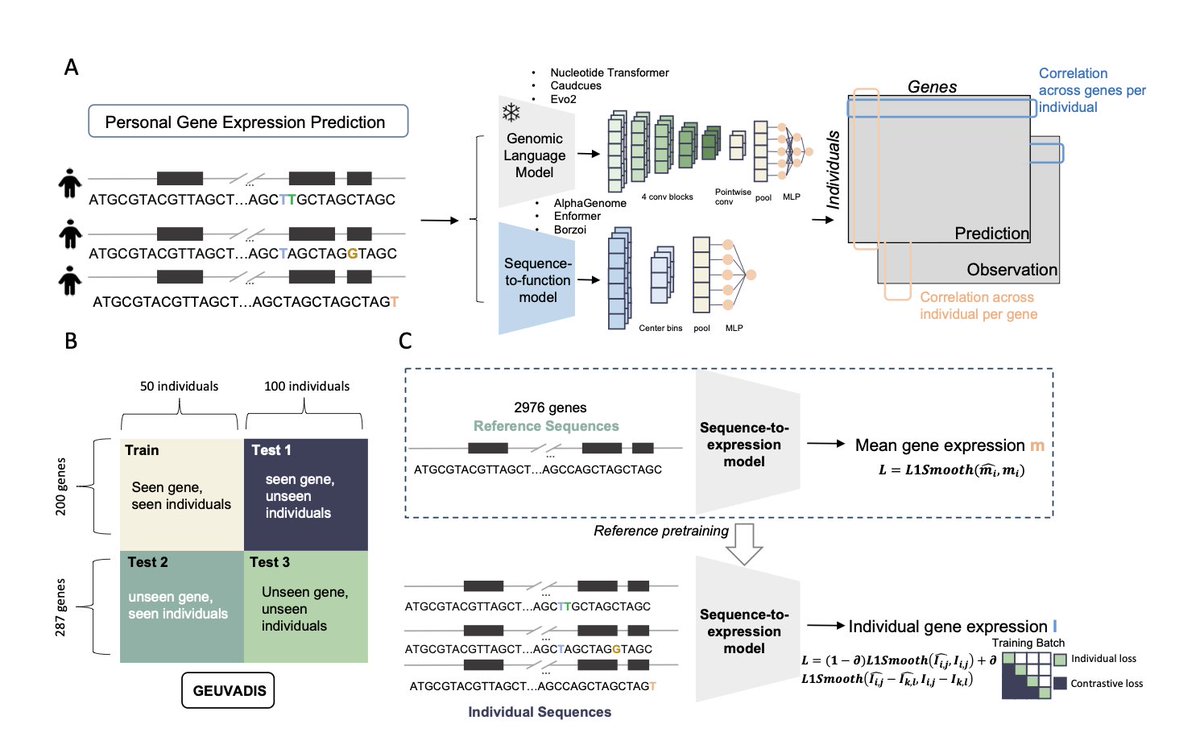
Assessing large-scale genomic language models in predicting personal gene expression: promises and limitations
1. A new study explores the potential and challenges of using genomic language models (gLMs) to predict individual gene expression. The research aims to evaluate how well these models can capture the variability in gene expression across different individuals, which is crucial for personalized genomics.
2. The study introduces a novel framework called gLM2X-Tower, designed to benchmark both gLMs and sequence-to-function (S2F) models on the task of predicting personal gene expression. This framework leverages paired personal genome-transcriptome data, allowing for a comprehensive assessment of model performance in different scenarios.
3. Key findings indicate that while gLMs and S2F models show promise in predicting gene expression for seen genes in new individuals, they struggle to generalize to unseen genes and individuals. This highlights a significant limitation in current models' ability to capture the complex regulatory mechanisms driving gene expression variability.
4. The research also demonstrates that incorporating personal genomes into the training process improves prediction accuracy for seen genes, suggesting that individual-level data can enhance model performance. However, this improvement does not extend to unseen contexts, indicating a need for more diverse training data and advanced model architectures.
5. The study emphasizes the importance of multi-species pretraining in gLMs, which outperformed human-only models in unseen contexts. This suggests that evolutionary diversity can provide valuable insights for improving gene expression prediction models.
6. Despite advancements in gLMs and S2F models, the study concludes that current approaches still face challenges in accurately predicting personal gene expression in unseen genomic sequences. Future work should focus on integrating more comprehensive training data and developing more efficient model architectures to overcome these limitations.
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/StatBiomed/gLM2X-…
#Genomics #PersonalizedMedicine #GenomicLanguageModels #GeneExpressionPrediction #ComputationalBiology
2
7
1,013
23 Mar 2025
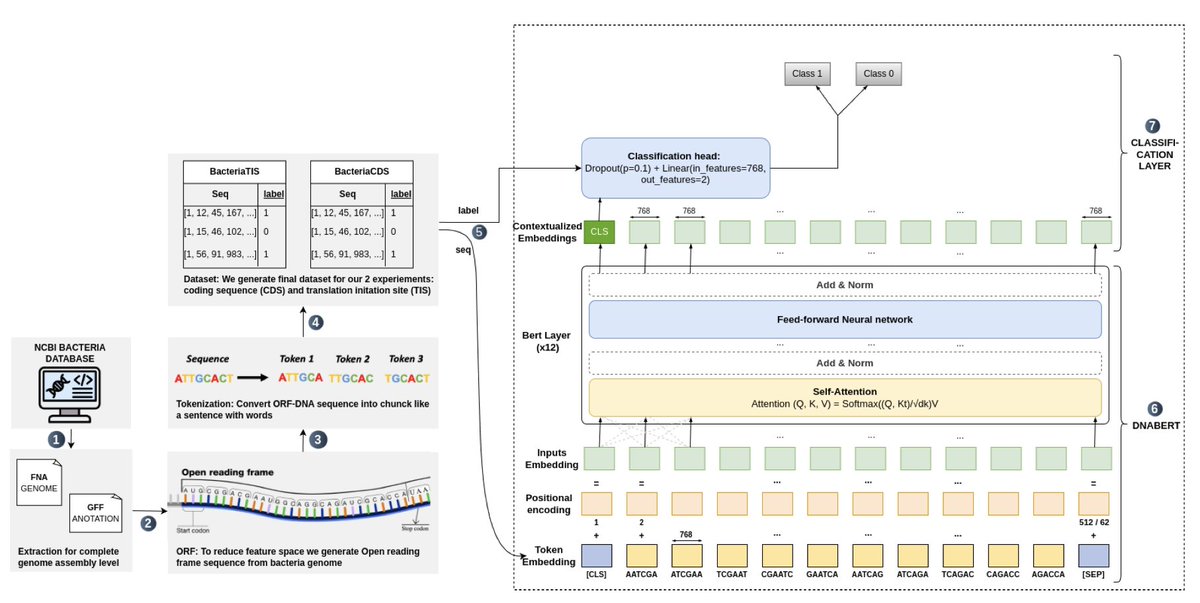
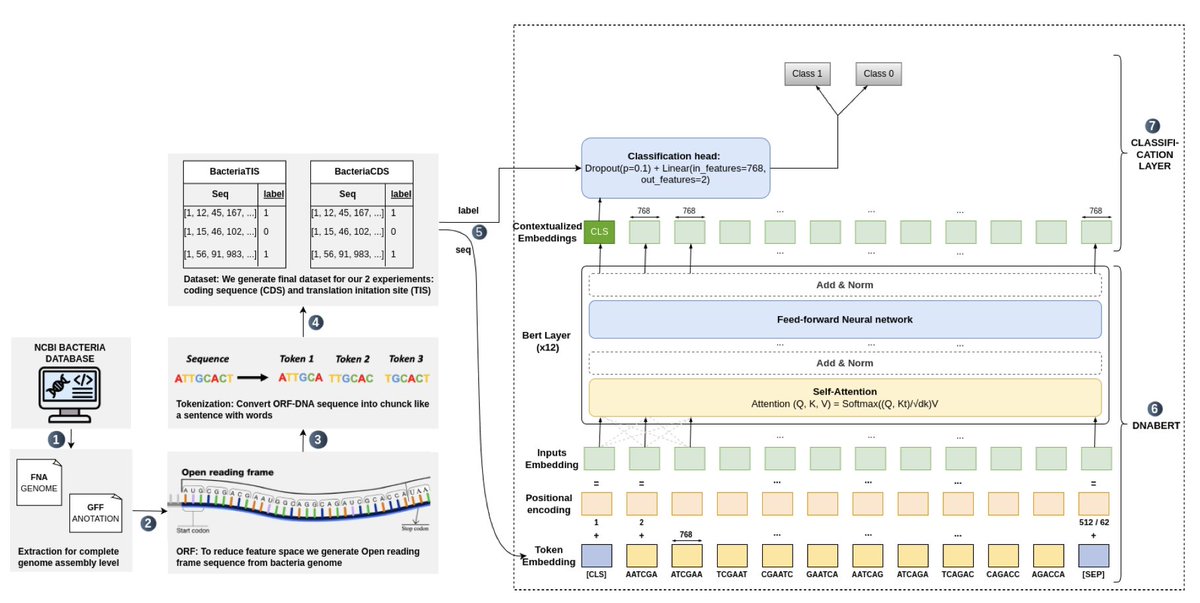
GENOMIC LANGUAGE MODELS (GLMS) DECODE BACTERIAL GENOMES FOR IMPROVED GENE PREDICTION AND TRANSLATION INITIATION SITE IDENTIFICATION
1. This paper introduces a transformer-based Genomic Language Model (gLM) called GeneLM, designed to improve gene prediction and Translation Initiation Site (TIS) identification in bacterial genomes. The approach draws inspiration from natural language processing models like BERT, applying them to genomic sequences.
2. GeneLM employs a two-stage framework: first, identifying Coding Sequence (CDS) regions, and then refining predictions by pinpointing the correct TIS within those regions. DNABERT, a specialized transformer architecture for DNA sequences, is fine-tuned using bacterial genomes from the NCBI database.
3. The model significantly improves gene prediction accuracy compared to conventional tools like Prodigal. Benchmarking results show that GeneLM reduces missed CDS predictions while increasing the number of correctly identified TIS, outperforming existing gene annotation methods.
4. The use of attention mechanisms within the transformer architecture allows GeneLM to capture meaningful biological patterns, such as upstream promoter regions relevant to TIS prediction. Visualizations show that the model effectively identifies biologically significant motifs across diverse bacterial species.
5. GeneLM demonstrates high generalization capability, achieving impressive results on unseen bacterial genomes. The study highlights the potential of using transformer-based models to enhance genome annotation, providing a more accurate tool for microbial genomic research.
6. Despite its performance, the authors acknowledge that transformer-based models require substantial computational resources and large-scale datasets, posing challenges for widespread adoption. Future work will focus on optimizing the model's efficiency and interpretability.
💻Code: github.com/Bioinformatics-UM…
📜Paper: biorxiv.org/content/10.1101/…
#GenomicLanguageModels #GenePrediction #Transformers #BacterialGenomics #MachineLearning #Bioinformatics #AI #GenomeAnnotation #DNABERT
4
7
1,668
23 Mar 2025
GENOMIC LANGUAGE MODELS (GLMS) DECODE BACTERIAL GENOMES FOR IMPROVED GENE PREDICTION AND TRANSLATION INITIATION SITE IDENTIFICATION
1. This paper introduces a transformer-based Genomic Language Model (gLM) called GeneLM, designed to improve gene prediction and Translation Initiation Site (TIS) identification in bacterial genomes. The approach draws inspiration from natural language processing models like BERT, applying them to genomic sequences.
2. GeneLM employs a two-stage framework: first, identifying Coding Sequence (CDS) regions, and then refining predictions by pinpointing the correct TIS within those regions. DNABERT, a specialized transformer architecture for DNA sequences, is fine-tuned using bacterial genomes from the NCBI database.
3. The model significantly improves gene prediction accuracy compared to conventional tools like Prodigal. Benchmarking results show that GeneLM reduces missed CDS predictions while increasing the number of correctly identified TIS, outperforming existing gene annotation methods.
4. The use of attention mechanisms within the transformer architecture allows GeneLM to capture meaningful biological patterns, such as upstream promoter regions relevant to TIS prediction. Visualizations show that the model effectively identifies biologically significant motifs across diverse bacterial species.
5. GeneLM demonstrates high generalization capability, achieving impressive results on unseen bacterial genomes. The study highlights the potential of using transformer-based models to enhance genome annotation, providing a more accurate tool for microbial genomic research.
6. Despite its performance, the authors acknowledge that transformer-based models require substantial computational resources and large-scale datasets, posing challenges for widespread adoption. Future work will focus on optimizing the model's efficiency and interpretability.
💻Code: github.com/Bioinformatics-UM…
📜Paper: biorxiv.org/content/10.1101/…
#GenomicLanguageModels #GenePrediction #Transformers #BacterialGenomics #MachineLearning #Bioinformatics #AI #GenomeAnnotation #DNABERT
3
3
1,107
9 Dec 2024
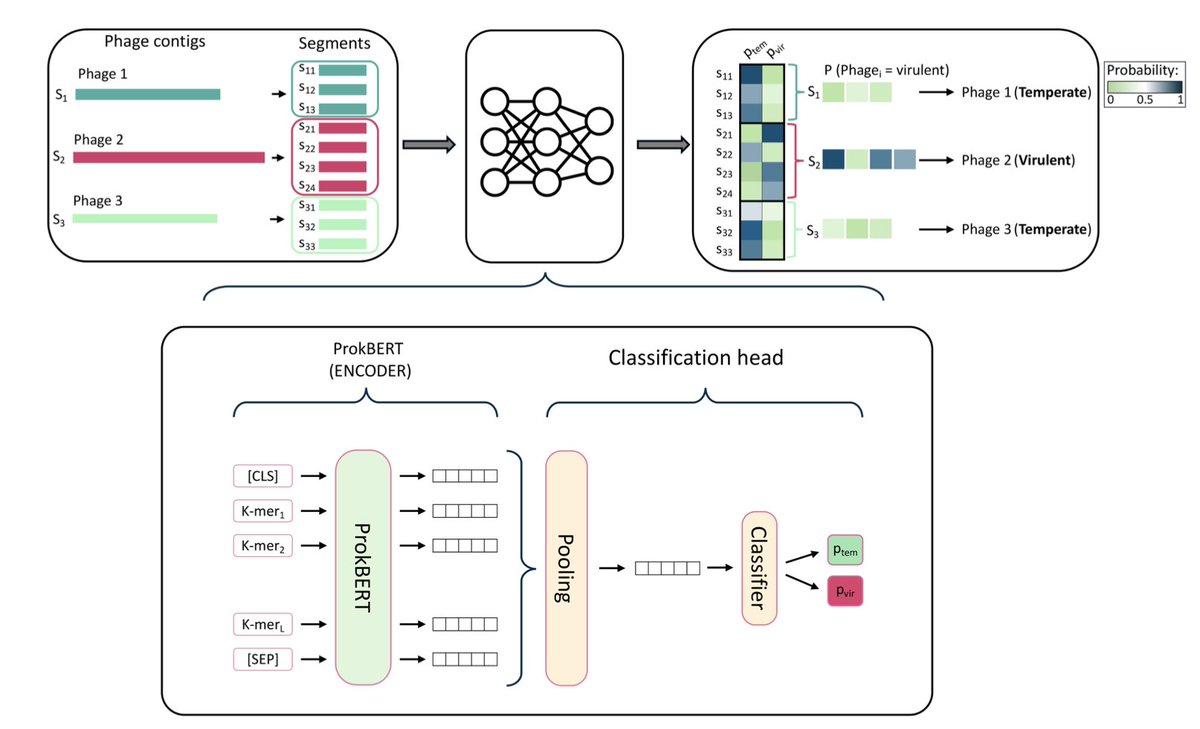
ProkBERT PhaStyle: Accurate Phage Lifestyle Prediction with Pretrained Genomic Language Models
1. Introduced ProkBERT PhaStyle, a fine-tuned genomic language model achieving state-of-the-art performance in classifying phage lifestyles (virulent vs. temperate) directly from nucleotide sequences without requiring protein annotations or complex pipelines.
2. ProkBERT PhaStyle consistently outperformed existing tools (BACPHLIP, PhaTYP, DeePhage) on fragmented and unseen phage sequences, achieving a balanced accuracy of 0.94 on 1022 bp fragments and generalizing well to extreme environmental datasets.
3. Leveraged innovative k-mer-based tokenization and compact architectures (~20M parameters) to deliver high accuracy with computational efficiency, making it scalable and suitable for large-scale metagenomic datasets.
4. Demonstrated superior generalization by accurately classifying sequences from Escherichia phages and extremophile phages that were absent in training data, highlighting robustness against dataset bias.
5. Employed weighted voting for contig classification, combining segment predictions to achieve holistic insights into phage genomes, ensuring applicability in practical bioinformatics workflows.
6. Highlighted practical benefits, including rapid inference speeds (0.52 MB/sec) and lightweight resource requirements compared to larger models like DNABERT-2 and Nucleotide Transformer.
7. Positioned ProkBERT PhaStyle as a transformative tool for ecological and clinical applications, facilitating research in phage therapy, microbiome engineering, and viral ecology with streamlined workflows.
💻Code: github.com/nbrg-ppcu/PhaStyl…
📜Paper: biorxiv.org/content/10.1101/…
#PhageResearch #GenomicLanguageModels #Bioinformatics #DeepLearning #Microbiology #ViralEcology
3
8
1,130