
30 Apr 2025
Understanding Protein Language Model Scaling on Mutation Effect Prediction
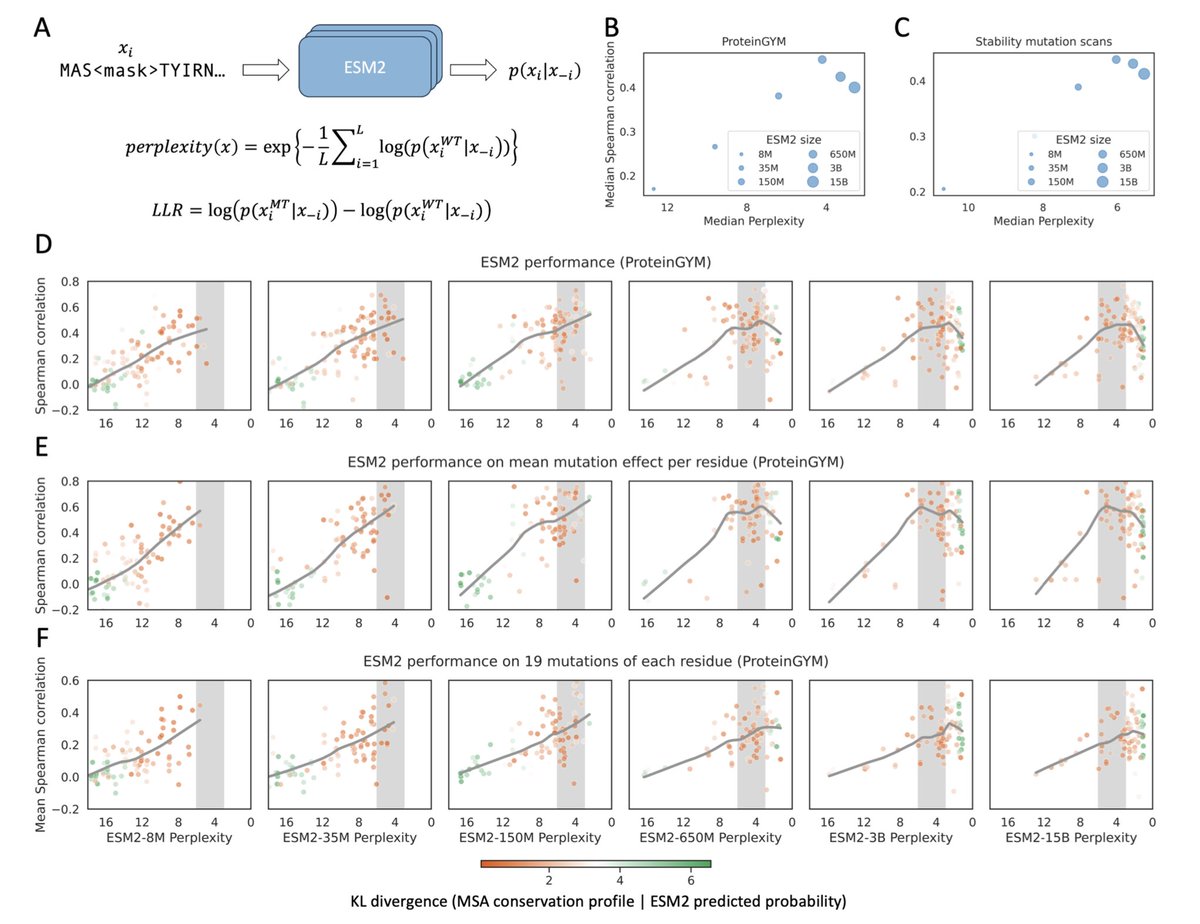
1. This study investigates why larger protein language models (pLMs), such as ESM2-3B and ESM2-15B, often underperform compared to medium-sized models like ESM2-650M in predicting mutation effects, despite achieving lower perplexity.
2. The authors show that mutation effect prediction performance peaks when model-predicted perplexity for a protein lies within the range of 3–6. Models outside this range tend to output indiscriminate log-likelihood ratios (LLRs), failing to distinguish deleterious from neutral mutations.
3. At low perplexity (typical for large models), LLRs are overly negative for most mutations, collapsing dynamic range. At high perplexity (typical for small models), LLRs cluster near zero. Both extremes diminish predictive resolution.
4. The study uses two large benchmarks—ProteinGYM and a mega-scale protein stability dataset—to show that model performance follows a rise-then-fall trend with respect to perplexity, robust across proteins with diverse sequence homology and structural contexts.
5. The authors link LLM-predicted amino acid distributions with conservation profiles from MSAs, finding highest agreement (lowest KL divergence) in the 3–6 perplexity range, where pLMs implicitly recapitulate evolutionary conservation.
6. Residue-level analyses reveal that low perplexity models lose specificity for contextually important positions, whereas optimal-perplexity models better capture both average residue fitness and substitution-specific effects.
7. Practical guidance is offered: if a model yields very low perplexity on a protein, switch to a smaller model; if perplexity is too high, fine-tune the model using homologous sequences to bring it into the optimal zone.
8. The study cautions that pLMs are not trained to model conservation but to predict wild-type residues, which can cause large models to overfit to dominant sequences, leading to poor mutation differentiation in highly conserved proteins.
9. Proteins outside natural evolutionary constraints—like viral or designed proteins—show lower performance even at ideal perplexity, indicating that pLMs may not generalize to synthetic biology without retraining on function-specific objectives.
10. The authors propose future training regimes that dynamically exclude overly predictable proteins during pLM training to optimize for mutation effect prediction, and advocate reconsidering perplexity as a universal benchmark for model quality.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLM #MutationEffect #Perplexity #ProteinEngineering #ComputationalBiology #ESM2 #ModelScaling #AI4Science #Bioinformatics
1
6
30
2,567
30 Apr 2025
Understanding Protein Language Model Scaling on Mutation Effect Prediction
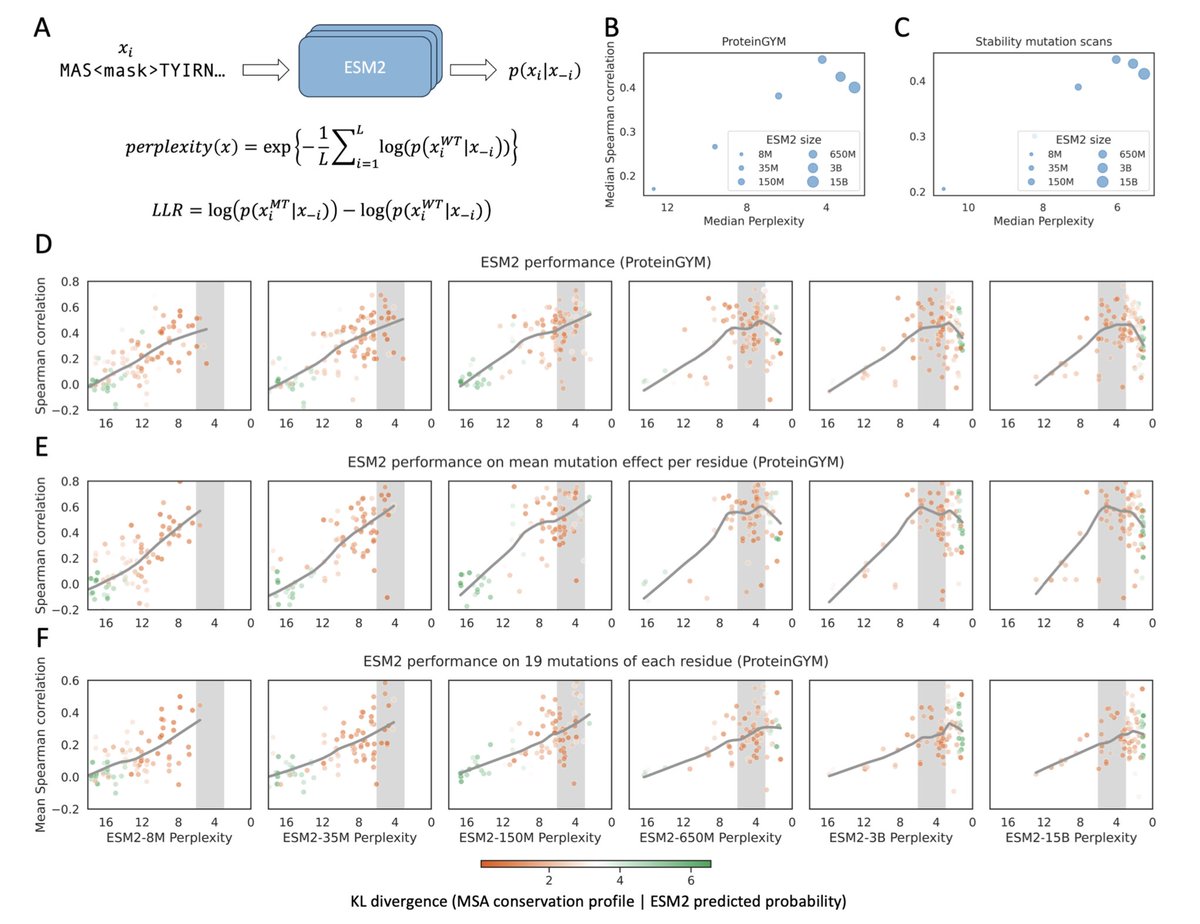
1. This study investigates why larger protein language models (pLMs), such as ESM2-3B and ESM2-15B, often underperform compared to medium-sized models like ESM2-650M in predicting mutation effects, despite achieving lower perplexity.
2. The authors show that mutation effect prediction performance peaks when model-predicted perplexity for a protein lies within the range of 3–6. Models outside this range tend to output indiscriminate log-likelihood ratios (LLRs), failing to distinguish deleterious from neutral mutations.
3. At low perplexity (typical for large models), LLRs are overly negative for most mutations, collapsing dynamic range. At high perplexity (typical for small models), LLRs cluster near zero. Both extremes diminish predictive resolution.
4. The study uses two large benchmarks—ProteinGYM and a mega-scale protein stability dataset—to show that model performance follows a rise-then-fall trend with respect to perplexity, robust across proteins with diverse sequence homology and structural contexts.
5. The authors link LLM-predicted amino acid distributions with conservation profiles from MSAs, finding highest agreement (lowest KL divergence) in the 3–6 perplexity range, where pLMs implicitly recapitulate evolutionary conservation.
6. Residue-level analyses reveal that low perplexity models lose specificity for contextually important positions, whereas optimal-perplexity models better capture both average residue fitness and substitution-specific effects.
7. Practical guidance is offered: if a model yields very low perplexity on a protein, switch to a smaller model; if perplexity is too high, fine-tune the model using homologous sequences to bring it into the optimal zone.
8. The study cautions that pLMs are not trained to model conservation but to predict wild-type residues, which can cause large models to overfit to dominant sequences, leading to poor mutation differentiation in highly conserved proteins.
9. Proteins outside natural evolutionary constraints—like viral or designed proteins—show lower performance even at ideal perplexity, indicating that pLMs may not generalize to synthetic biology without retraining on function-specific objectives.
10. The authors propose future training regimes that dynamically exclude overly predictable proteins during pLM training to optimize for mutation effect prediction, and advocate reconsidering perplexity as a universal benchmark for model quality.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLM #MutationEffect #Perplexity #ProteinEngineering #ComputationalBiology #ESM2 #ModelScaling #AI4Science #Bioinformatics
1
4
13
2,237
17 Feb 2025
Predicting Mutation-Induced Relative Protein-Ligand Binding Affinity Changes via Conformational Sampling and Diversity Integration with Subsampled Alphafold2 in Few-Shot Learning
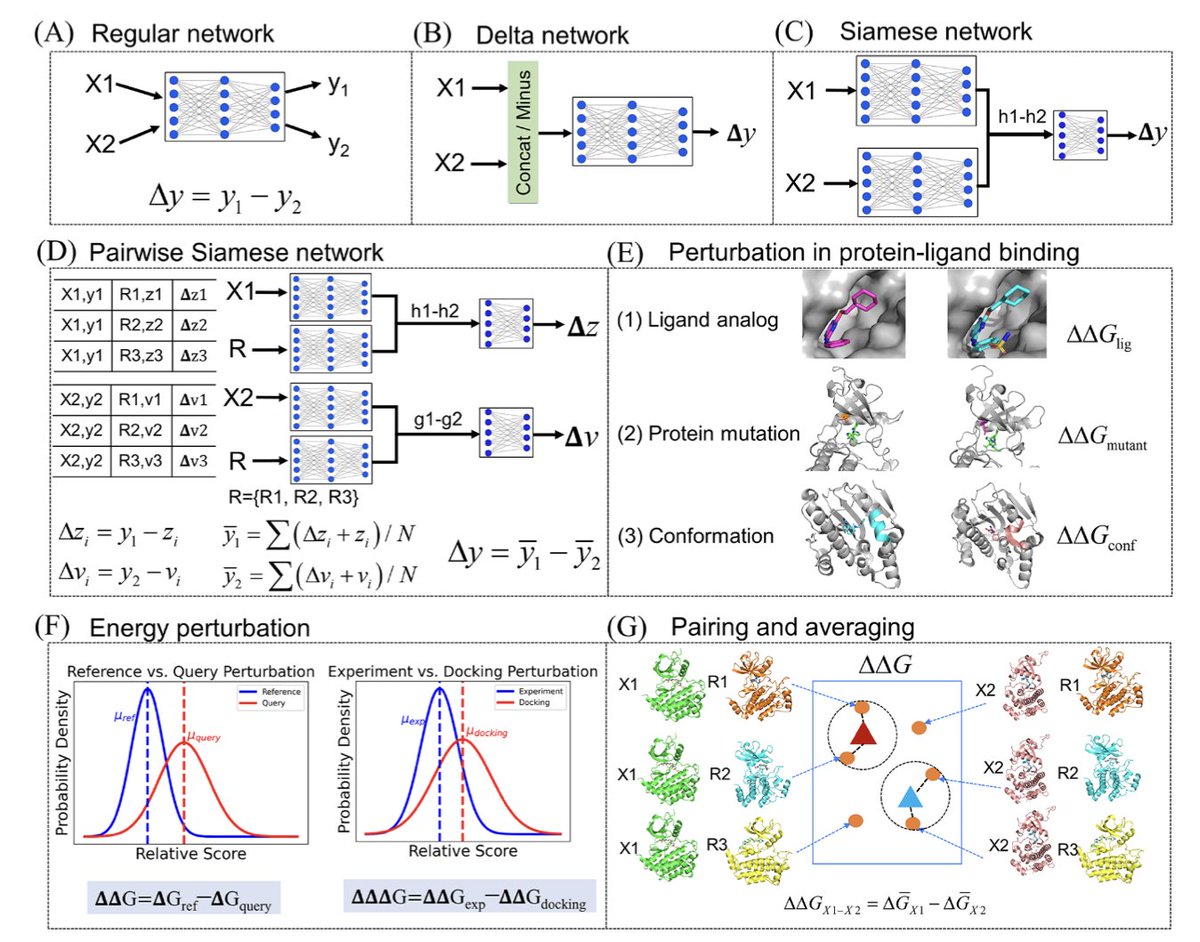
1/ This study introduces a powerful few-shot learning approach designed to predict mutation-induced changes in protein-ligand binding affinity. The method integrates subsampled AlphaFold2, ensemble docking, and Siamese learning for more accurate predictions of binding affinity alterations due to mutations.
2/ The authors utilize AlphaFold2 subsampling to generate diverse conformational ensembles for each protein variant, improving the model’s ability to handle the dynamic nature of proteins and avoid reliance on single static structures. This captures the conformational variability crucial for accurate affinity predictions.
3/ In addition to subsampling, the model incorporates Gaussian fitting to select the most likely conformational states, ensuring that the predicted binding energies are derived from the most relevant and representative structures, even for mutated proteins with limited available data.
4/ The core innovation of this work is the use of a Siamese network architecture to predict relative binding affinity changes. By comparing pairs of protein conformations, the network effectively learns the differences in affinity induced by mutations and ligand variations, rather than focusing on absolute values.
5/ The proposed STGNet model (Structure and Graph-aware Siamese Network) incorporates protein contact maps, molecular graphs, and protein-ligand interaction graphs, using advanced message-passing neural networks (MPNN) to handle these multimodal inputs and extract critical structural and interaction features.
6/ This framework is validated through benchmarking on the Tyrosine kinase inhibitor (TKI) dataset, demonstrating significant improvements in predictive accuracy over traditional methods like molecular docking and free energy perturbation (FEP ), particularly in predicting mutation-induced changes in binding affinity.
7/ The study shows that using data augmentation strategies, specifically expanding datasets through the inclusion of multiple conformational states, helps the model generalize better. A 50-fold data expansion achieves optimal prediction accuracy across a range of TKIs.
8/ This research has important implications for drug discovery, particularly in understanding how mutations affect drug resistance. By incorporating structural diversity and conformation-based energy differences, the model offers a robust tool for predicting drug efficacy and resistance profiles.
💻Code: github.com/AIMedDrug/STGNet
📜Paper: biorxiv.org/content/10.1101/…
#MachineLearning #DrugDiscovery #ComputationalBiology #ProteinBinding #MutationEffect #AlphaFold2 #ProteinLigandInteraction #SiameseNetwork #Bioinformatics #DeepLearning #DrugResistance
10
52
2,612
17 Feb 2025
Predicting Mutation-Induced Relative Protein-Ligand Binding Affinity Changes via Conformational Sampling and Diversity Integration with Subsampled Alphafold2 in Few-Shot Learning
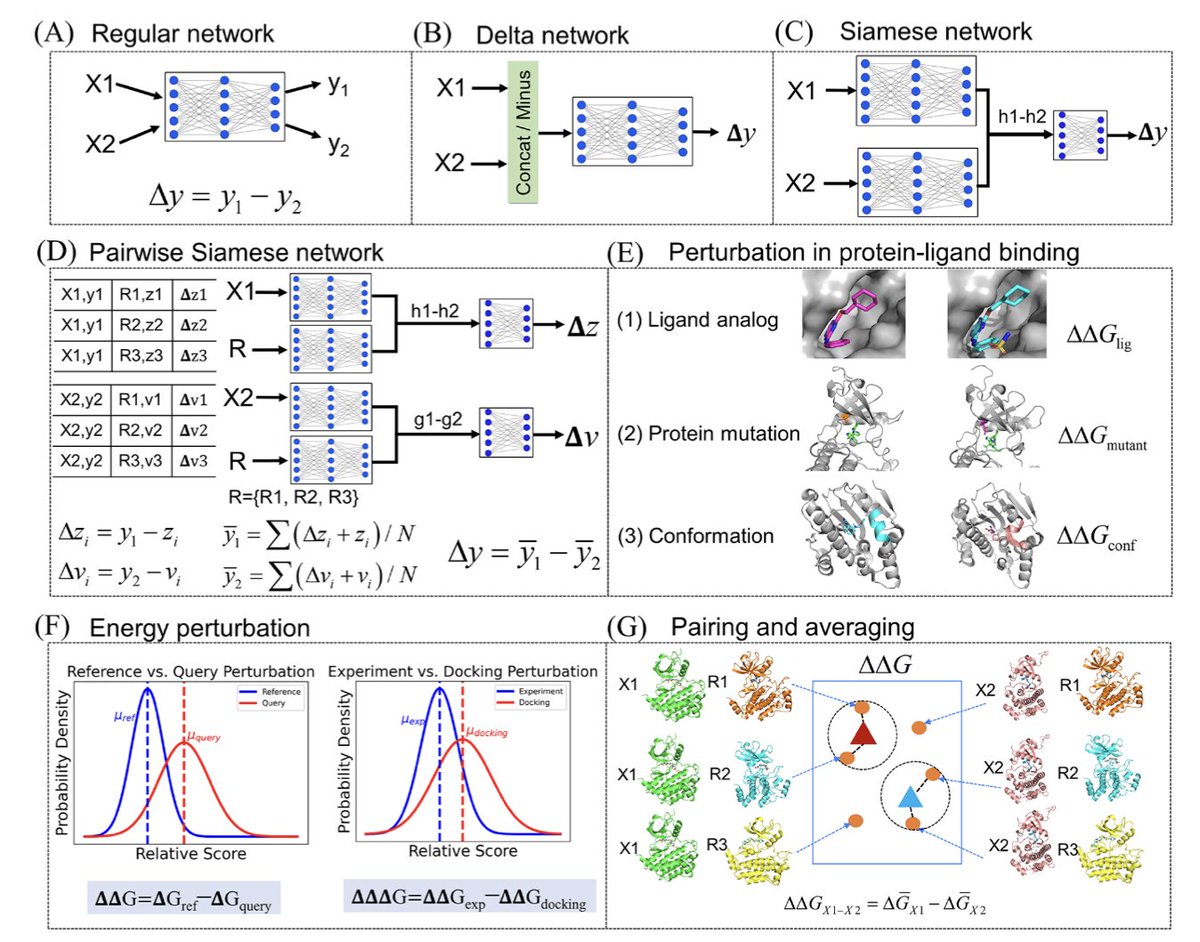
1/ This study introduces a powerful few-shot learning approach designed to predict mutation-induced changes in protein-ligand binding affinity. The method integrates subsampled AlphaFold2, ensemble docking, and Siamese learning for more accurate predictions of binding affinity alterations due to mutations.
2/ The authors utilize AlphaFold2 subsampling to generate diverse conformational ensembles for each protein variant, improving the model’s ability to handle the dynamic nature of proteins and avoid reliance on single static structures. This captures the conformational variability crucial for accurate affinity predictions.
3/ In addition to subsampling, the model incorporates Gaussian fitting to select the most likely conformational states, ensuring that the predicted binding energies are derived from the most relevant and representative structures, even for mutated proteins with limited available data.
4/ The core innovation of this work is the use of a Siamese network architecture to predict relative binding affinity changes. By comparing pairs of protein conformations, the network effectively learns the differences in affinity induced by mutations and ligand variations, rather than focusing on absolute values.
5/ The proposed STGNet model (Structure and Graph-aware Siamese Network) incorporates protein contact maps, molecular graphs, and protein-ligand interaction graphs, using advanced message-passing neural networks (MPNN) to handle these multimodal inputs and extract critical structural and interaction features.
6/ This framework is validated through benchmarking on the Tyrosine kinase inhibitor (TKI) dataset, demonstrating significant improvements in predictive accuracy over traditional methods like molecular docking and free energy perturbation (FEP ), particularly in predicting mutation-induced changes in binding affinity.
7/ The study shows that using data augmentation strategies, specifically expanding datasets through the inclusion of multiple conformational states, helps the model generalize better. A 50-fold data expansion achieves optimal prediction accuracy across a range of TKIs.
8/ This research has important implications for drug discovery, particularly in understanding how mutations affect drug resistance. By incorporating structural diversity and conformation-based energy differences, the model offers a robust tool for predicting drug efficacy and resistance profiles.
💻Code: github.com/AIMedDrug/STGNet
📜Paper: biorxiv.org/content/10.1101/…
#MachineLearning #DrugDiscovery #ComputationalBiology #ProteinBinding #MutationEffect #AlphaFold2 #ProteinLigandInteraction #SiameseNetwork #Bioinformatics #DeepLearning #DrugResistance
3
11
1,077

25 Mar 2024
It was truly a pleasure to speak on #AI for #Innovation, #Genomics, and #Medicine at our @MIT #IAP class with @RickardGabriels on #FoundationModels and #GenerativeAI.
So many extraordinary advances to cover, each more exciting than the other.
Video here: youtube.com/watch?v=pHzdw6YW…
Topics touched upon:
- #CNNs for #RegulatoryGenomics and #MutationEffect prediction
- #GNNs for #DrugDevelopment
- #Transformers for #ProteinFolding
- #VAEs for #SingleCell expression
- #CLIP for #MedicalImaging-#Legends
- #MultiModal function-sequence-structure for #TherapeuticDesign
- #CognitiveSpace navigation for #patients #EHR, #ProteinFunction, #CellState, #DrugEffects, #ScientificPapers, #Grants, #Patents, #Education, #Investment, #Discovery, #Management, #Productivity, and #Innovation.
Some slides attached below, lots more coming! :-)




25 Mar 2024

🧬 Honored to have Professor @manoliskellis guest lecture at MIT's course on Foundation Models & Generative AI. This session delves into the exciting interface of AI, Biology & Genomics, a field seeing incredible advancements. It's a must-see: youtu.be/pHzdw6YWB9I
2
22
138
19,239