
🧠 AI scaling is entering a new phase. DeepSeek’s mHC architecture shows how smarter model design, not just bigger compute, can unlock stability and efficiency in 2026.
Read the full insight 👉 1950.ai/post/from-resnet-to-…
#AIArchitecture #ModelScaling #AdvancedAI #1950ai
1
4
591

30 Dec 2025
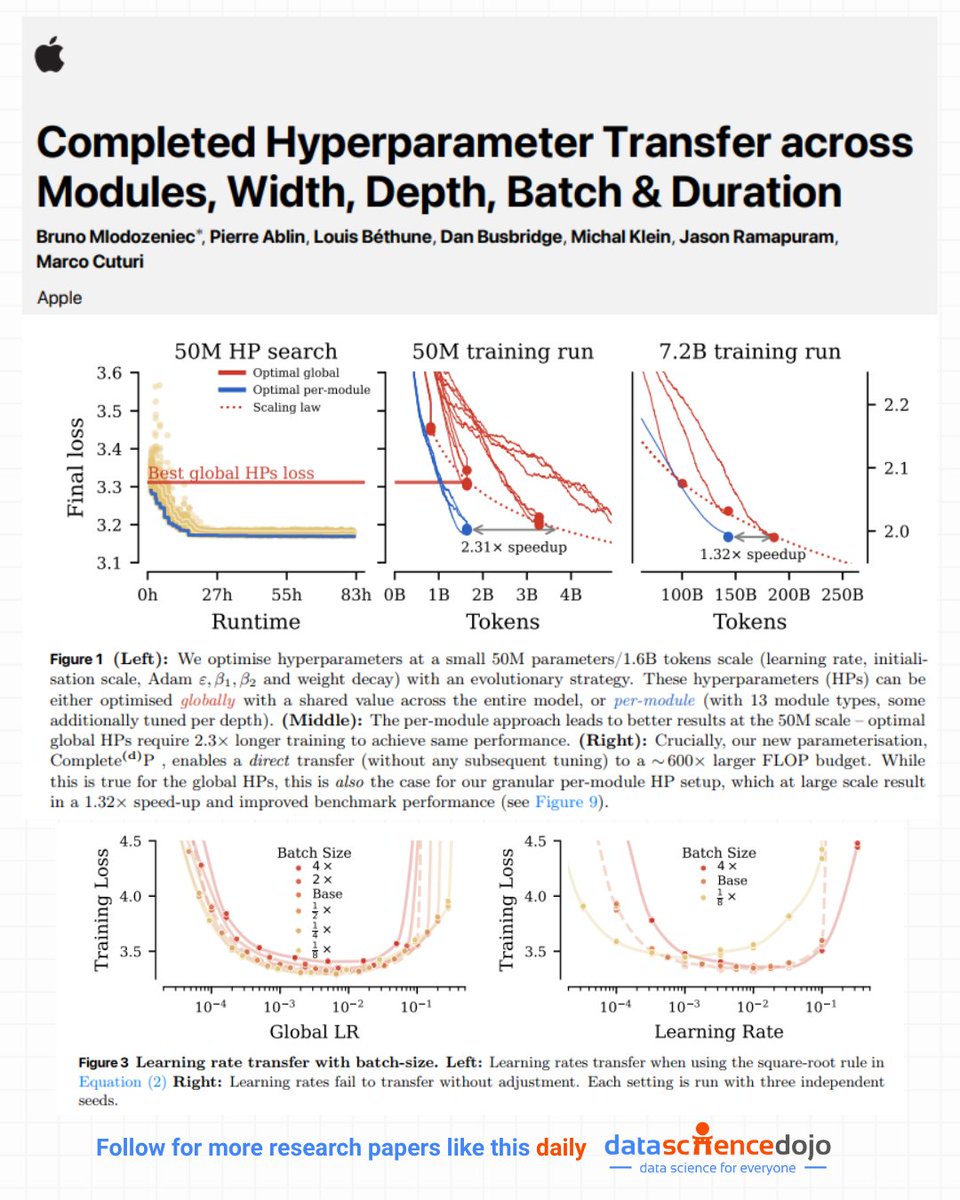
📢 Apple just released a paper that tackles one of the most persistent practical challenges in training large models: hyperparameter tuning at scale.
While many advances in deep learning focus on bigger architectures or more data, this work dives deep into a deceptively difficult problem: how do you find good hyperparameters — like learning rates, weight decay, and optimizer settings — once you scale models up by orders of magnitude?
The authors build on recent ideas in hyperparameter parameterizations and extend them with a new framework called Complete(d)P, which unifies scaling across model width, depth, batch size, and training duration. Instead of treating each scaling axis separately, their approach lets you search for optimal hyperparameters on a small model and then transfer them reliably to much larger models — even when you change batch size or the number of training tokens.
A key insight from this paper is that tuning hyperparameters at scale doesn’t have to mean expensive grid searches or manual trial-and-error on every new configuration. With the right parameterization, the structure of the optimization landscape can be understood well enough at small scale that the same settings still work when everything grows — reducing training cost and improving stability across scales.
The authors also show that this per-module hyperparameter transfer works better than global tuning alone, and that it can yield real speedups and more reliable training behavior as models get larger.
In short, this paper is a thoughtful reminder that scaling ML systems isn’t just about bigger models — it’s about smarter training design. And that optimizing how we train at scale can unlock efficiency gains that are just as important as any architectural breakthrough.
#MachineLearning #HyperparameterTuning #ModelScaling #AITraining #DeepLearning #Optimization #Research #LLMs #EfficientAI
1
4
928

25 Nov 2025
Diversity Beats Size Scaling for Chemical Language Models
1. A new study challenges the notion that bigger is always better in chemical language models (CLMs). Researchers found that beyond a certain threshold, increasing model size does not improve hit generation rates in drug design tasks. Instead, dataset diversification significantly boosts hit diversity with minimal impact on hit rates.
2. The study systematically evaluated encoder–decoder transformers trained on paired textual molecular representations. It revealed that while model scaling has diminishing returns, dataset scaling also provides limited gains after a certain point. The focus should shift from sheer scale to curated and diverse data.
3. A key innovation is the introduction of a dataset diversification strategy. By clustering molecules based on molecular weight and then sampling from these clusters, researchers created datasets that yielded more diverse hits without compromising on the number of hits generated. This approach could transform how training data is curated for CLMs.
4. The findings have practical implications for drug discovery. They suggest that investing in data curation and diversity may be more impactful than continuously scaling up model size or dataset volume. This could lead to more efficient and innovative molecular design processes in the pharmaceutical industry.
5. The study highlights the importance of evaluating models directly on downstream tasks rather than relying on proxy measures like pre-training loss. This approach provides a more accurate assessment of model performance in real-world drug design scenarios.
📜Paper: doi.org/10.26434/chemrxiv-20…
#ChemicalLanguageModels #DrugDiscovery #AIinPharma #DatasetDiversity #ModelScaling
1
5
13
1,062

22 Oct 2025
Forget about genshin powerscaling. Genshin modelscaling. They're all being height crept. What did they feed varka to build him Like That
1
7
315
30 Apr 2025
Understanding Protein Language Model Scaling on Mutation Effect Prediction
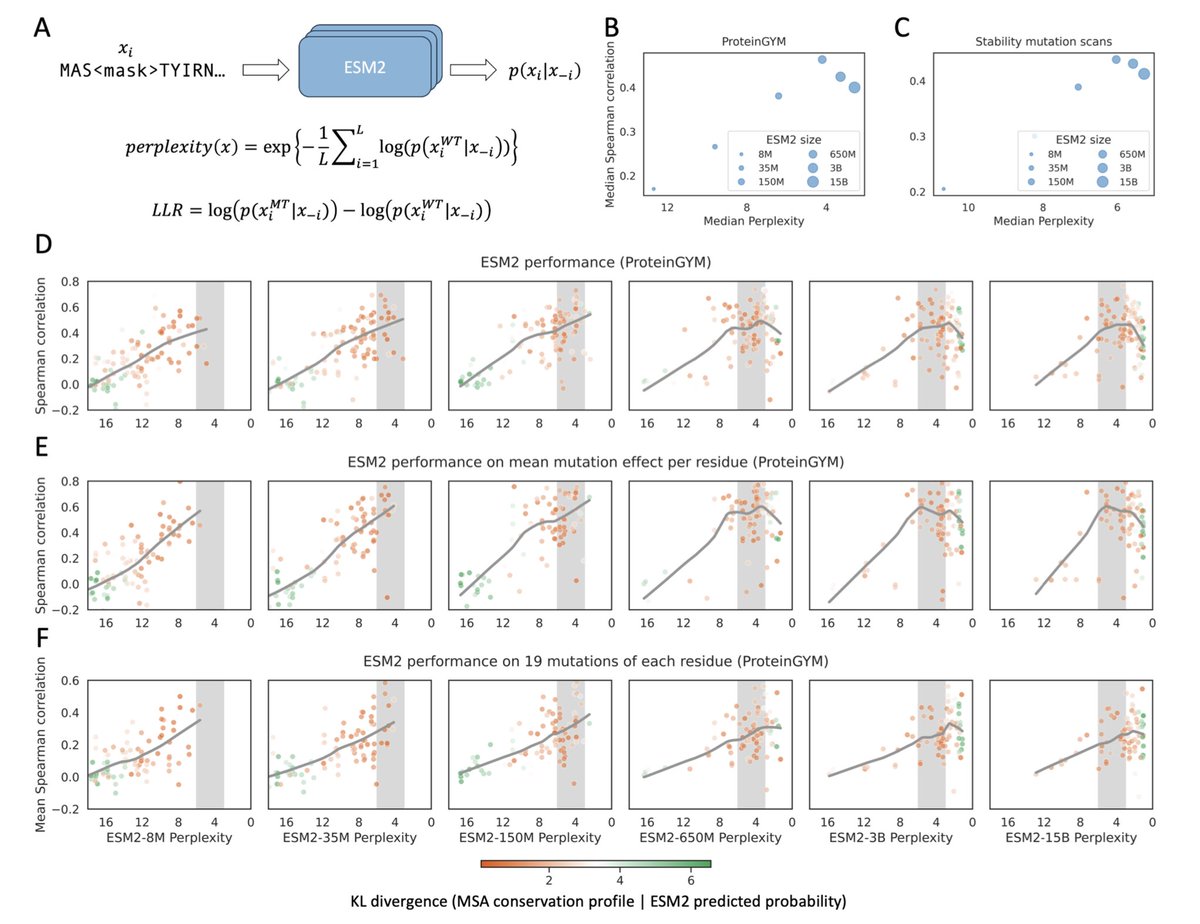
1. This study investigates why larger protein language models (pLMs), such as ESM2-3B and ESM2-15B, often underperform compared to medium-sized models like ESM2-650M in predicting mutation effects, despite achieving lower perplexity.
2. The authors show that mutation effect prediction performance peaks when model-predicted perplexity for a protein lies within the range of 3–6. Models outside this range tend to output indiscriminate log-likelihood ratios (LLRs), failing to distinguish deleterious from neutral mutations.
3. At low perplexity (typical for large models), LLRs are overly negative for most mutations, collapsing dynamic range. At high perplexity (typical for small models), LLRs cluster near zero. Both extremes diminish predictive resolution.
4. The study uses two large benchmarks—ProteinGYM and a mega-scale protein stability dataset—to show that model performance follows a rise-then-fall trend with respect to perplexity, robust across proteins with diverse sequence homology and structural contexts.
5. The authors link LLM-predicted amino acid distributions with conservation profiles from MSAs, finding highest agreement (lowest KL divergence) in the 3–6 perplexity range, where pLMs implicitly recapitulate evolutionary conservation.
6. Residue-level analyses reveal that low perplexity models lose specificity for contextually important positions, whereas optimal-perplexity models better capture both average residue fitness and substitution-specific effects.
7. Practical guidance is offered: if a model yields very low perplexity on a protein, switch to a smaller model; if perplexity is too high, fine-tune the model using homologous sequences to bring it into the optimal zone.
8. The study cautions that pLMs are not trained to model conservation but to predict wild-type residues, which can cause large models to overfit to dominant sequences, leading to poor mutation differentiation in highly conserved proteins.
9. Proteins outside natural evolutionary constraints—like viral or designed proteins—show lower performance even at ideal perplexity, indicating that pLMs may not generalize to synthetic biology without retraining on function-specific objectives.
10. The authors propose future training regimes that dynamically exclude overly predictable proteins during pLM training to optimize for mutation effect prediction, and advocate reconsidering perplexity as a universal benchmark for model quality.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLM #MutationEffect #Perplexity #ProteinEngineering #ComputationalBiology #ESM2 #ModelScaling #AI4Science #Bioinformatics
1
6
30
2,567
30 Apr 2025
Understanding Protein Language Model Scaling on Mutation Effect Prediction
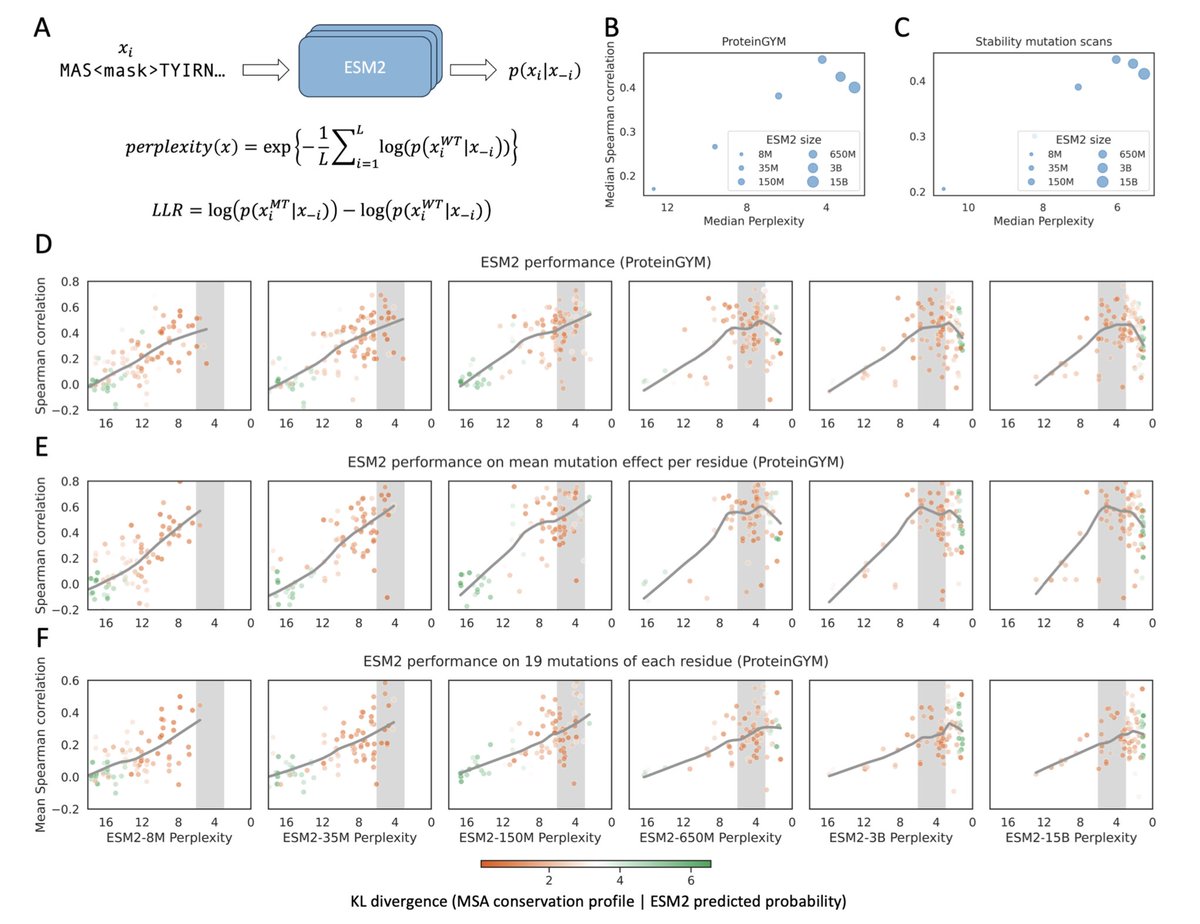
1. This study investigates why larger protein language models (pLMs), such as ESM2-3B and ESM2-15B, often underperform compared to medium-sized models like ESM2-650M in predicting mutation effects, despite achieving lower perplexity.
2. The authors show that mutation effect prediction performance peaks when model-predicted perplexity for a protein lies within the range of 3–6. Models outside this range tend to output indiscriminate log-likelihood ratios (LLRs), failing to distinguish deleterious from neutral mutations.
3. At low perplexity (typical for large models), LLRs are overly negative for most mutations, collapsing dynamic range. At high perplexity (typical for small models), LLRs cluster near zero. Both extremes diminish predictive resolution.
4. The study uses two large benchmarks—ProteinGYM and a mega-scale protein stability dataset—to show that model performance follows a rise-then-fall trend with respect to perplexity, robust across proteins with diverse sequence homology and structural contexts.
5. The authors link LLM-predicted amino acid distributions with conservation profiles from MSAs, finding highest agreement (lowest KL divergence) in the 3–6 perplexity range, where pLMs implicitly recapitulate evolutionary conservation.
6. Residue-level analyses reveal that low perplexity models lose specificity for contextually important positions, whereas optimal-perplexity models better capture both average residue fitness and substitution-specific effects.
7. Practical guidance is offered: if a model yields very low perplexity on a protein, switch to a smaller model; if perplexity is too high, fine-tune the model using homologous sequences to bring it into the optimal zone.
8. The study cautions that pLMs are not trained to model conservation but to predict wild-type residues, which can cause large models to overfit to dominant sequences, leading to poor mutation differentiation in highly conserved proteins.
9. Proteins outside natural evolutionary constraints—like viral or designed proteins—show lower performance even at ideal perplexity, indicating that pLMs may not generalize to synthetic biology without retraining on function-specific objectives.
10. The authors propose future training regimes that dynamically exclude overly predictable proteins during pLM training to optimize for mutation effect prediction, and advocate reconsidering perplexity as a universal benchmark for model quality.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLM #MutationEffect #Perplexity #ProteinEngineering #ComputationalBiology #ESM2 #ModelScaling #AI4Science #Bioinformatics
1
4
13
2,237

2 Apr 2024
[1/n]
🎉🎉🎉 Excited to share our latest work: "The Fine Line: Navigating Large Language Model Pretraining with Down-streaming Capability Analysis"! We delve into the dynamics of LLMs across different scales and domains.
💡Highlights include:
🗺️ Comprehensive Model Evaluation: Leveraging an array of LLMs (Baichuan-7B, DeepSeek-7B, Amber-7B, OpenLLaMA-7B, Yi-34B, DeepSeek-67B) for extensive downstream task assessment, we illuminate diverse performance landscapes and emergent capabilities, charting new courses for model development.
📈 Task Dynamic Prediction: We've found that a model's performance on known tasks can predict its success on similar, unseen tasks. A leap towards understanding LLMs' learning process!
🌱 Emergent Synergies & Skill Evolution: Insights from one domain can fuel learning in another, mimicking human cognitive growth and suggesting a curriculum for model training. At the same time, we trace the unique timelines of emergent skills across models, showcasing the complex journey of AI learning and adaptation.
🔧 Impact of Training Strategies: Analysis of 7b-scale models reveals the significant role of dataset quality, learning rate, and architecture in early-stage training efficiency.
🧠 Model Scale & Reasoning Tasks: Larger models excel in reasoning, but smart strategies can boost smaller models to compete.
📏 Reevaluating Scaling Laws: Our findings challenge and extend the traditional scaling laws linking training data size to LLM performance on downstream tasks. It's not just about more data; it's about smarter use leading to transformative results.
We're also releasing intermediate checkpoints for Amber-7B and OpenLLaMA-7B to foster further research! 🌟
arxiv.org/pdf/2404.01204.pdf Dive in to explore how these insights can reshape your strategies for developing foundational models. 🚀🌍💡#AIResearch #LargeLanguageModels #ScalingLaw #DeepLearning #ModelScaling #EmergentCapabilities #TrainingStrategies #InnovationInAI

2
29
101
24,324

30 Nov 2019
Discount Code: ADVENT20
Promotion period only valid from 30.11.2019 to 01.12.2019 - Promotion valid on selected articles. Only while stocks last. Not combinable with other promotions.
To the Revell Online Shop: fcld.ly/u8xvwiz
#giftset #modelscaling #scalemodels

1
6

12 Jun 2019
EfficientNet: Improving Accuracy and Efficiency through AutoML and Model Scaling bit.ly/2RakTIQ #google #googleAI #AI #RT #efficientnet #autoML #ML #AI #MachineLearning #ModelScaling #nasnet #google
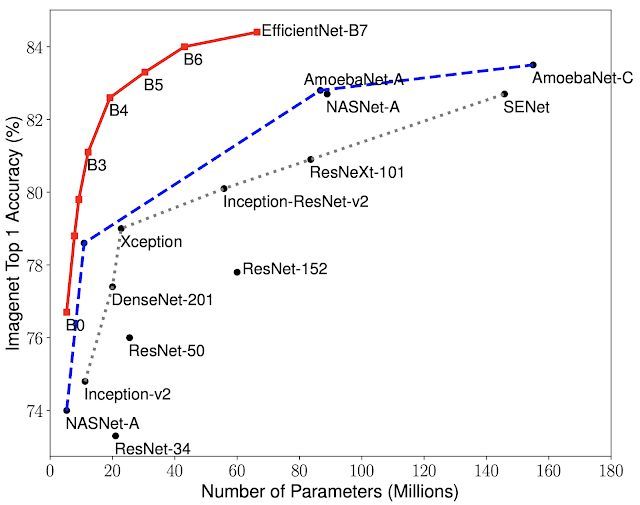
1
