Protein language models trained on biophysical dynamics inform mutation effects @PNASNews
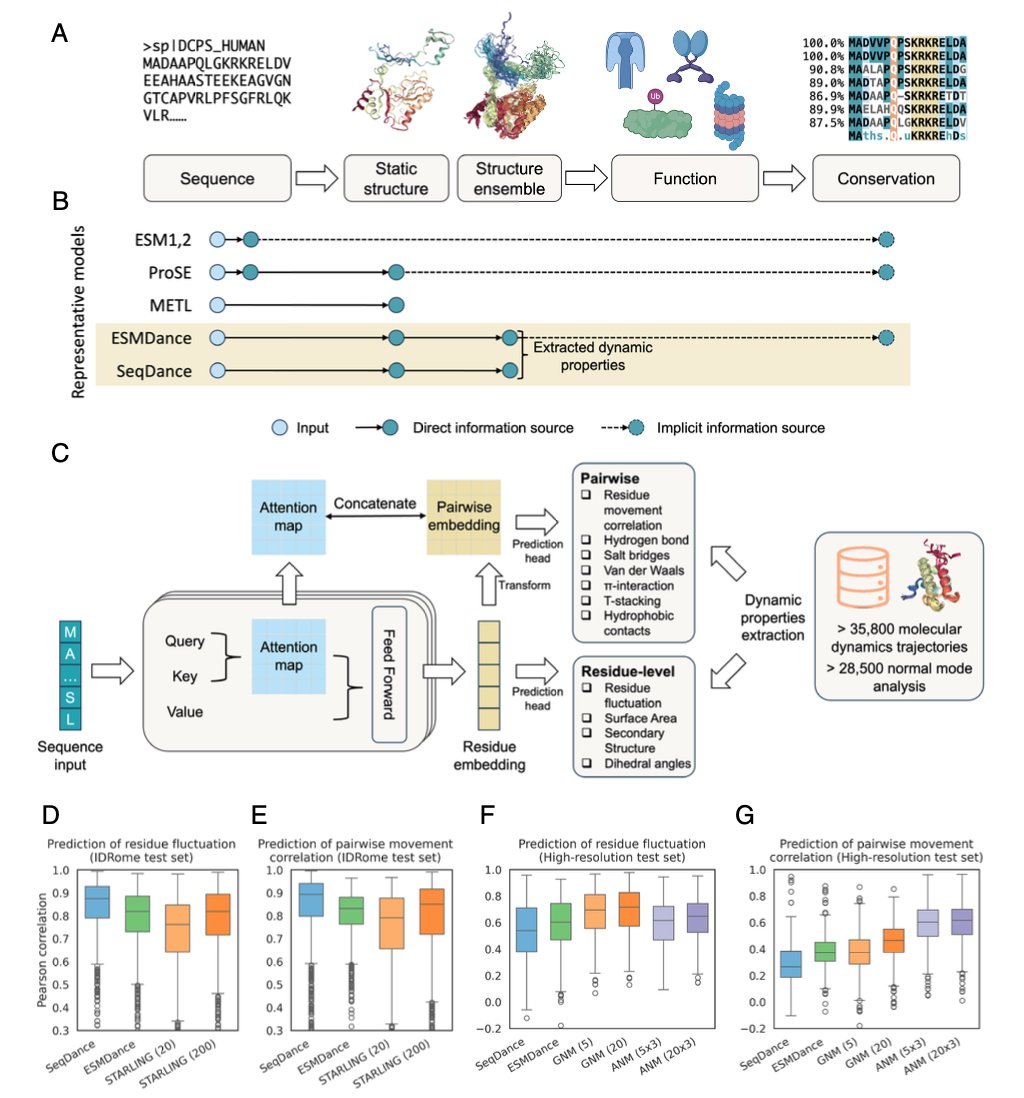
1. Researchers have developed two novel protein language models, SeqDance and ESMDance, which integrate dynamic biophysical properties derived from molecular dynamics simulations and normal mode analyses to predict protein behavior and mutation effects. These models address the limitations of existing deep learning models that rely predominantly on static structures and sequences.
2. SeqDance, trained from scratch on dynamic properties, demonstrates superior performance in capturing local dynamic interactions and residue comovement, especially for intrinsically disordered proteins (IDRs). Its attention mechanism effectively captures dynamic interactions directly from sequence data without relying on evolutionary information.
3. ESMDance, built upon the ESM2 model, leverages both evolutionary and dynamic information to significantly outperform existing models in predicting mutation effects on protein stability. This model shows robust generalization, particularly for designed and viral proteins where evolutionary information is sparse.
4. The study highlights the importance of incorporating protein dynamics into language models, as it enables more accurate predictions of conformational properties and mutation effects compared to models relying solely on static structures or evolutionary data. This approach could bridge the gap between sequence, dynamics, and function in computational protein design.
5. The models were trained on a large dataset of over 64,000 proteins, including both high-resolution MD simulation trajectories and low-resolution data from normal mode analysis. This comprehensive dataset allowed the models to learn a wide range of dynamic properties, enhancing their applicability across diverse protein types.
6. SeqDance's embeddings were shown to encode global protein conformation properties, allowing for accurate predictions of end-to-end distance, asphericity, and radius of gyration for both ordered and disordered proteins. This demonstrates the model's ability to generalize beyond the training tasks.
7. The research also evaluated the models' performance on predicting mutation effects without specific training on mutation data. ESMDance achieved a median Spearman correlation of 0.46 across 412 proteins, outperforming both SeqDance and ESM2 models in zero-shot prediction.
📜Paper: pnas.org/doi/10.1073/pnas.25…
#ProteinLanguageModels #Biophysics #MolecularDynamics #ProteinDynamics #MutationEffects #ComputationalBiology
1
22
115
6,113

1 Dec 2025
DeepPni: Language and graph-based model for mutation-driven protein nucleic acid energetics
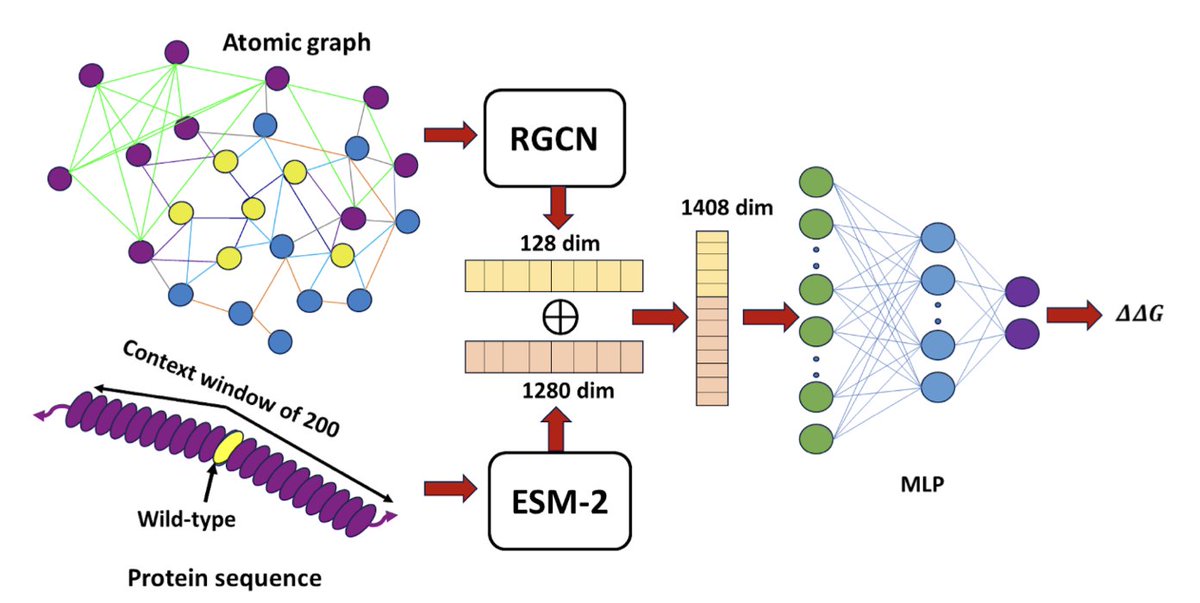
1. DeepPni is a novel deep learning framework that integrates graph-based structural representations with ESM-2 protein sequence embeddings to predict mutation-induced binding free energy changes in protein-nucleic acid complexes. This approach leverages both spatial and sequential information to achieve superior predictive performance.
2. The model incorporates an edge-aware relational graph convolutional network (RGCN) to encode complex structural features around the mutation site, capturing the heterogeneous interactions between proteins and nucleic acids. This is combined with ESM-2 embeddings to provide a comprehensive representation of the mutational context.
3. DeepPni demonstrates robust performance across diverse datasets, achieving a Pearson correlation coefficient (PCC) of 0.76 on a large dataset of 1951 mutations. It consistently outperforms existing methods, including DeePnaP and MutPni, across various benchmarks.
4. The study highlights the importance of relational features in the atomic graph, showing that incorporating edge-specific information significantly improves prediction accuracy. This innovation sets DeepPni apart from traditional models that use simpler graph representations.
5. DeepPni maintains stable performance across different experimental conditions, including protein-DNA and protein-RNA complexes, as well as various temperature splits. This versatility makes it a valuable tool for analyzing the effects of mutations on protein-nucleic acid interactions.
6. External validation on the ProNab dataset further confirms DeepPni's generalizability, with high correlation values for different experimental methodologies (ITC, GS, FP, FB). This suggests the model's applicability in real-world scenarios.
📜Paper: arxiv.org/abs/2511.22239
#DeepLearning #ProteinNucleicAcidInteractions #MutationEffects #ComputationalBiology #Bioinformatics
1
3
15
1,404

5 Oct 2025
Twin Peaks: Dual-Head Architecture for Structure-Free Prediction of Protein-Protein Binding Affinity and Mutation Effects
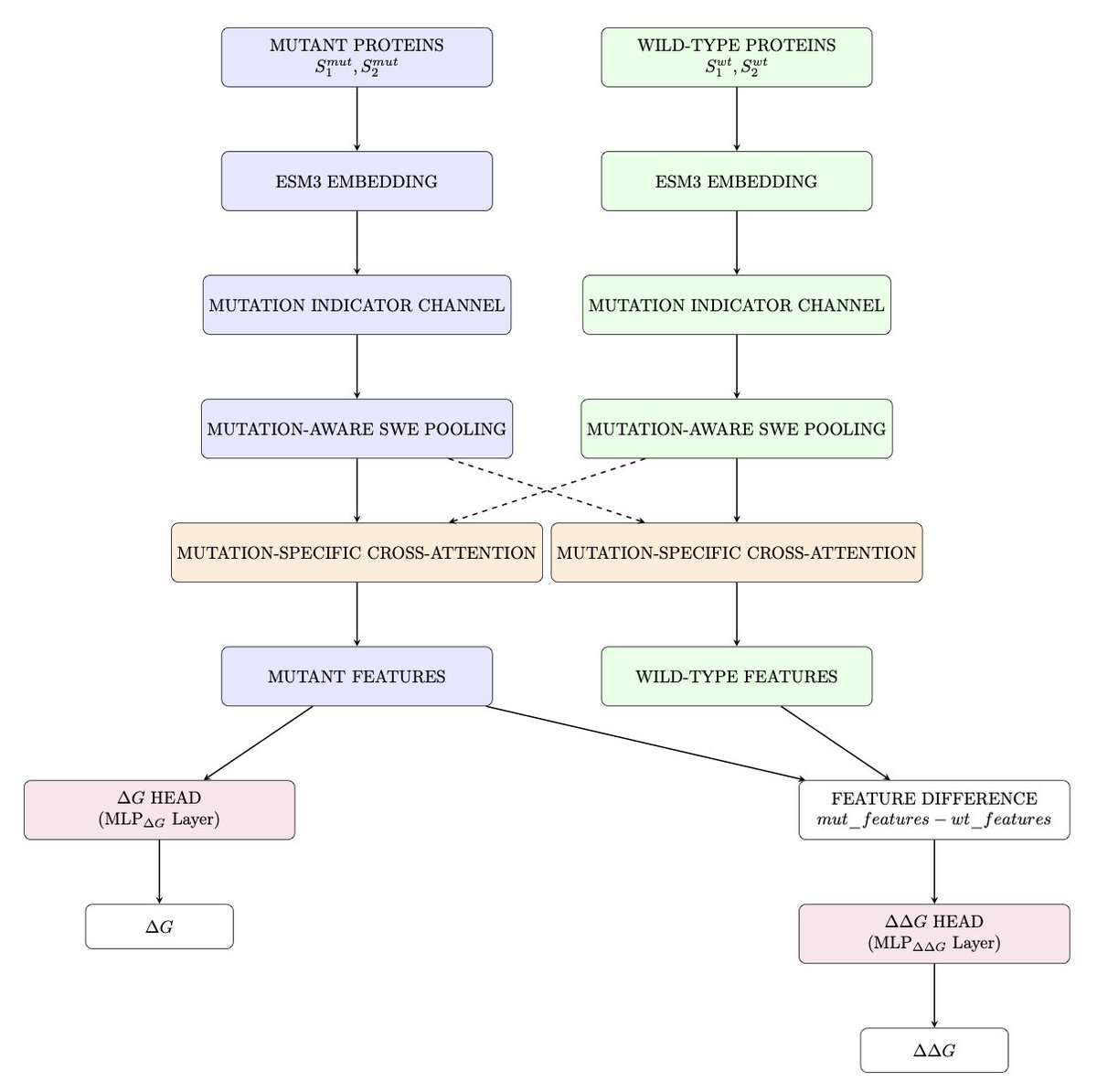
1. The article introduces "Twin Peaks," a novel dual-head deep learning architecture that simultaneously predicts protein-protein binding affinity (∆G) and mutation-induced affinity changes (∆∆G) using only protein sequence information. This innovation eliminates the need for structural data, a significant advancement in the field of computational biology.
2. The model employs a specialized mutation-aware embedding strategy and a novel cross-attention mechanism to detect and amplify signals from critical hotspot residues that contribute to binding affinity changes. This approach is particularly effective for proteins with unknown structures or those that are difficult to crystallize, such as viral and intrinsically disordered proteins.
3. The architecture combines transformer-based encoders with a learnable sliced window embedding layer to manage variable-length sequences efficiently. It also uses a multi-layer transformer encoder with bidirectional self-attention to capture intra-protein patterns and cross-attention layers to model interactions between protein pairs.
4. The dual-head prediction network allows for task-specific optimization while leveraging common features. This design ensures robust generalization and prevents information leakage between training and validation sets. The model achieves a ∆∆G validation Pearson correlation of 0.485 and a ∆G Pearson correlation of 0.638.
5. The study integrates complementary datasets from SKEMPI v2 and PDBbind with a rigorous protein domain-based splitting strategy. This approach yields 6,732 training samples across 2,322 unique protein complexes and 856 validation samples from 140 unique complexes, all from non-overlapping protein families.
6. The authors highlight that existing approaches often fail to generalize outside their training distribution, but Twin Peaks maintains consistent performance across diverse protein families, including those absent from the training data. This capability is crucial for real-world applications where structural similarity cannot be guaranteed.
7. The model's ability to detect the effects of single residue mutations without structural information is a significant advancement. This sensitivity arises from the mutation-aware components that explicitly track mutation positions and their influence throughout the network.
8. Twin Peaks enables rapid screening of thousands of potential mutations in minutes, a stark contrast to the hours or days required by structure-based methods. This speed and efficiency make it a valuable tool for drug discovery, protein engineering, and understanding disease mechanisms.
📜Paper: arxiv.org/abs/2509.22950v1
#ProteinBinding #MutationEffects #DeepLearning #ComputationalBiology #StructuralBiology #ProteinEngineering #DrugDiscovery
3
15
1,503
27 Aug 2025
A Practical Guide to Transition State Analysis in Biomolecular Simulations with TS-DAR
1. This guide introduces TS-DAR, a novel computational framework for identifying transition states in biomolecular conformational changes. Transition states are critical yet sparse conformations that define rate-limiting steps in molecular processes, and TS-DAR offers a robust solution to detect them systematically.
2. TS-DAR leverages a deep learning model to map protein conformations from molecular dynamics (MD) simulations onto a hyperspherical latent space. This low-dimensional representation retains essential kinetic information while allowing for the automated identification of transition states through a combination of VAMP-2 and dispersion loss functions.
3. The framework is particularly innovative in its use of out-of-distribution (OOD) detection. By treating transition states as OOD relative to metastable states, TS-DAR can identify these rare conformations that are crucial for understanding biomolecular mechanisms and developing targeted therapeutics.
4. The tutorial provides a step-by-step workflow for implementing TS-DAR, including MD sampling, featurization, model training, and MSM construction. It also offers practical advice on hyperparameter tuning and model evaluation, making it accessible for researchers in computational biophysics.
5. The efficacy of TS-DAR is demonstrated across multiple systems, from simple 2D potentials to complex biomolecules like the DNA motor protein AlkD. It outperforms previous methods in both accuracy and efficiency, uncovering novel insights into key interactions that enable proteins to overcome free energy barriers.
6. The guide includes detailed examples for alanine dipeptide, the villin headpiece (HP35), and protein phosphatase 2A (PP2A). These examples illustrate how TS-DAR can be applied to different systems, highlighting its versatility and potential for advancing studies on drug binding, enzyme activity, and mutation effects.
7. The integration of TS-DAR with MSMs allows for accurate kinetic modeling of biomolecular systems. The tutorial explains how to construct an MSM from TS-DAR-derived states and validate it using the Chapman-Kolmogorov test, ensuring that the model captures the essential long-timescale kinetics.
8. Future perspectives for improving TS-DAR are also discussed, such as incorporating equivariant neural networks for more efficient feature selection and using TS-DAR-derived collective variables for enhanced sampling techniques like metadynamics.
📜Paper: doi.org/10.26434/chemrxiv-20…
#BiomolecularSimulations #TransitionStateAnalysis #DeepLearning #ComputationalBiophysics #ProteinDynamics #DrugDiscovery #EnzymeActivity #MutationEffects

1
3
18
1,363
30 May 2025
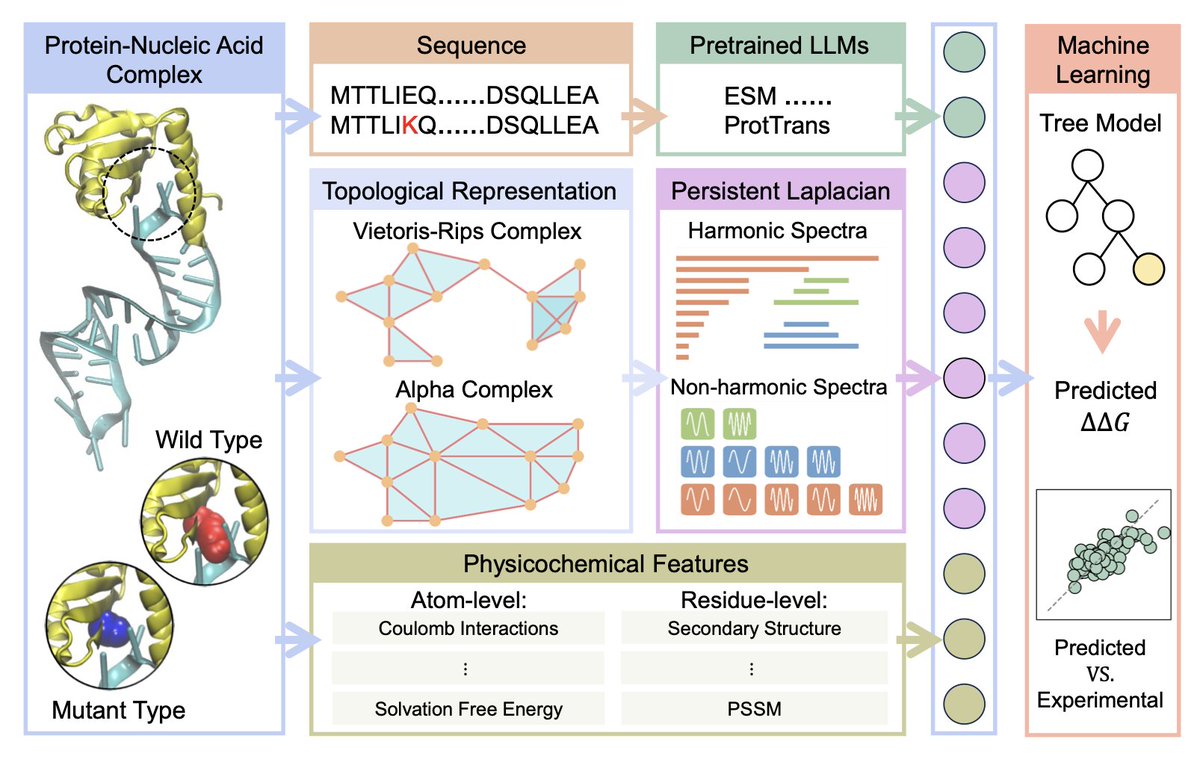
Topological Machine Learning for Protein-Nucleic Acid Binding Affinity Changes Upon Mutation
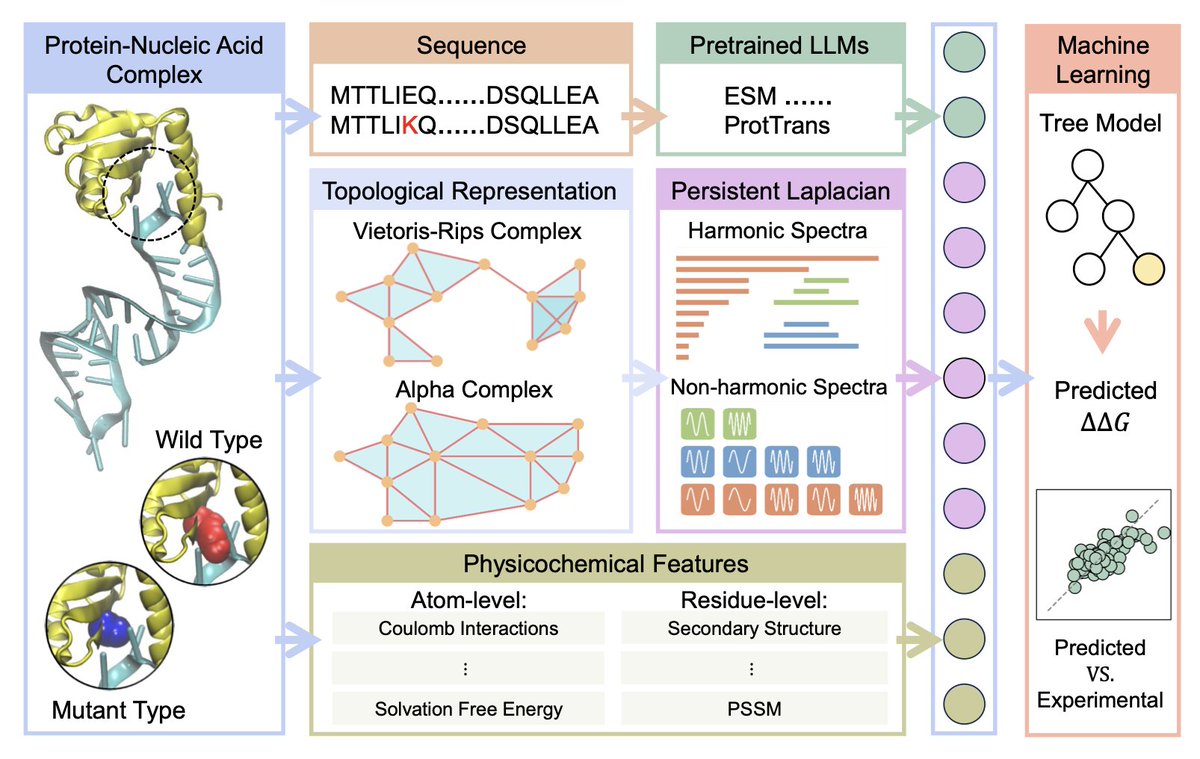
1.This study introduces TopoML, a new topological machine learning model that predicts how single-point mutations affect protein–nucleic acid binding affinities. It is the first to apply persistent Laplacian-based features to this domain and achieves state-of-the-art performance on both protein-DNA and protein-RNA datasets.
2.TopoML integrates three complementary feature types: persistent Laplacian-based topological features, physicochemical descriptors at both atom and residue levels, and protein sequence embeddings from a pretrained Transformer model (ESM-2). This multi-view representation captures both structural and sequence-level nuances of the binding interface.
3.For protein-RNA interactions, TopoML achieves a Pearson correlation coefficient (PCC) of 0.72 and MAE of 0.77 kcal/mol, outperforming prior leading models like PRA-MutPred (PCC 0.67). It also surpasses energy-based baselines PEMPNI and PNBACE in benchmark settings.
4.On protein-DNA interactions, TopoML also leads the field: a 10-fold cross-validation yields a PCC of 0.681 and MAE of 0.612 kcal/mol, outperforming SAMPDI-3Dv2 (PCC 0.65). When trained and evaluated on standard benchmark splits, it consistently delivers improved prediction accuracy.
5.Persistent Laplacians encode both topological (harmonic) and geometric (non-harmonic) properties of simplicial complexes formed at the mutation and binding regions. This richer representation outperforms classic persistent homology across many tasks and is central to the model’s predictive power.
6.The topological features alone provide strong performance—on protein-DNA interactions, using only topological features yields PCC 0.648, higher than using physicochemical or sequence features alone. This emphasizes the value of persistent Laplacian structures for modeling mutation-induced changes.
7.The model uses a gradient boosting tree for regression, combining the diverse features effectively. While this choice balances performance and interpretability, the authors suggest future exploration of ensemble and deep learning methods to further enhance accuracy.
8.Evaluation across mutation types (hydrophobic, polar, charged, alanine) and structural regions (core, surface, rim) reveals that TopoML maintains robust predictive performance. However, prediction accuracy slightly drops for underrepresented mutation types like positively charged residues, indicating areas for dataset expansion.
9.Interestingly, the model captures biophysically meaningful trends—for example, all alanine substitutions tend to increase ∆∆G, reflecting loss of favorable interactions. This alignment with known molecular principles supports the model's reliability.
10.TopoML's architecture and feature extraction pipeline are transparent and reproducible. The authors provide detailed procedures, from dataset curation and mutation modeling to topological complex construction and embedding computation.
11.The study opens promising directions for future research, including the use of persistent sheaf Laplacians or Dirac operators, and ensembling multiple learners. The current results demonstrate the untapped potential of topological approaches in understanding mutation impacts beyond protein-protein systems.
💻Code: github.com/LiuXiangMath/Topo…
📜Paper: arxiv.org/abs/2505.22786v1
#ProteinDesign #MutationEffects #BindingAffinity #MachineLearning #TopologicalDataAnalysis #Bioinformatics #StructuralBiology #PersistentLaplacian #ProteinDNA #ProteinRNA
3
13
796
30 May 2025
Topological Machine Learning for Protein-Nucleic Acid Binding Affinity Changes Upon Mutation
1.This study introduces TopoML, a new topological machine learning model that predicts how single-point mutations affect protein–nucleic acid binding affinities. It is the first to apply persistent Laplacian-based features to this domain and achieves state-of-the-art performance on both protein-DNA and protein-RNA datasets.
2.TopoML integrates three complementary feature types: persistent Laplacian-based topological features, physicochemical descriptors at both atom and residue levels, and protein sequence embeddings from a pretrained Transformer model (ESM-2). This multi-view representation captures both structural and sequence-level nuances of the binding interface.
3.For protein-RNA interactions, TopoML achieves a Pearson correlation coefficient (PCC) of 0.72 and MAE of 0.77 kcal/mol, outperforming prior leading models like PRA-MutPred (PCC 0.67). It also surpasses energy-based baselines PEMPNI and PNBACE in benchmark settings.
4.On protein-DNA interactions, TopoML also leads the field: a 10-fold cross-validation yields a PCC of 0.681 and MAE of 0.612 kcal/mol, outperforming SAMPDI-3Dv2 (PCC 0.65). When trained and evaluated on standard benchmark splits, it consistently delivers improved prediction accuracy.
5.Persistent Laplacians encode both topological (harmonic) and geometric (non-harmonic) properties of simplicial complexes formed at the mutation and binding regions. This richer representation outperforms classic persistent homology across many tasks and is central to the model’s predictive power.
6.The topological features alone provide strong performance—on protein-DNA interactions, using only topological features yields PCC 0.648, higher than using physicochemical or sequence features alone. This emphasizes the value of persistent Laplacian structures for modeling mutation-induced changes.
7.The model uses a gradient boosting tree for regression, combining the diverse features effectively. While this choice balances performance and interpretability, the authors suggest future exploration of ensemble and deep learning methods to further enhance accuracy.
8.Evaluation across mutation types (hydrophobic, polar, charged, alanine) and structural regions (core, surface, rim) reveals that TopoML maintains robust predictive performance. However, prediction accuracy slightly drops for underrepresented mutation types like positively charged residues, indicating areas for dataset expansion.
9.Interestingly, the model captures biophysically meaningful trends—for example, all alanine substitutions tend to increase ∆∆G, reflecting loss of favorable interactions. This alignment with known molecular principles supports the model's reliability.
10.TopoML's architecture and feature extraction pipeline are transparent and reproducible. The authors provide detailed procedures, from dataset curation and mutation modeling to topological complex construction and embedding computation.
11.The study opens promising directions for future research, including the use of persistent sheaf Laplacians or Dirac operators, and ensembling multiple learners. The current results demonstrate the untapped potential of topological approaches in understanding mutation impacts beyond protein-protein systems.
💻Code:
github.com/LiuXiangMath/Topo…
📜Paper:
arxiv.org/abs/2505.22786v1
#ProteinDesign #MutationEffects #BindingAffinity #MachineLearning #TopologicalDataAnalysis #Bioinformatics #StructuralBiology #PersistentLaplacian #ProteinDNA #ProteinRNA
3
9
796
7 Apr 2025
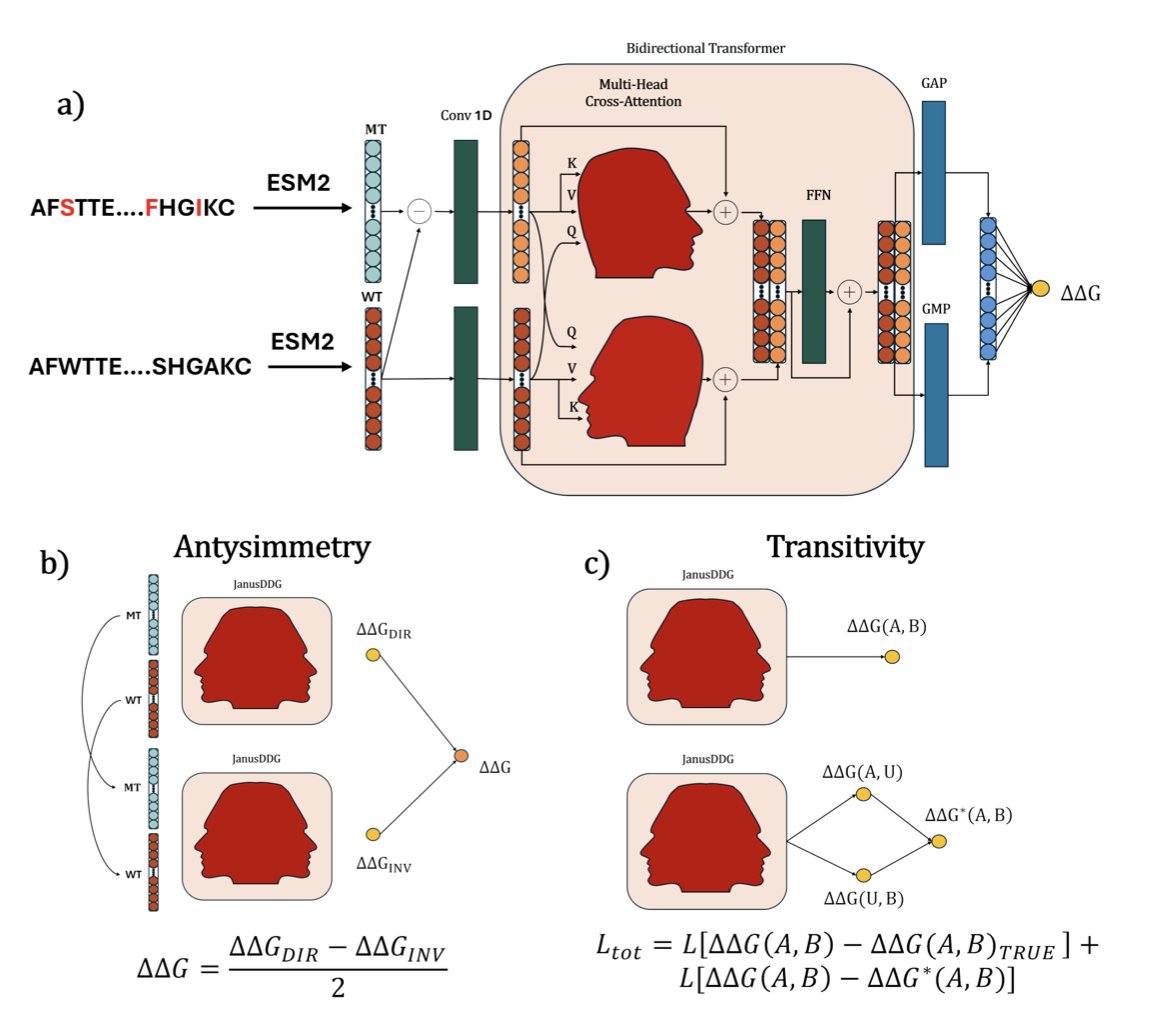
JanusDDG: A Thermodynamics-Compliant Model for Sequence-Based Protein Stability via Two-Fronts Multi-Head Attention
1. JanusDDG is a new sequence-based model that predicts changes in protein stability (ΔΔG) upon mutation, while strictly adhering to thermodynamic principles such as antisymmetry and transitivity.
2. Unlike conventional models that rely on 3D structures, JanusDDG predicts ΔΔG directly from amino acid sequences, making it broadly applicable and computationally efficient.
3. The model introduces a novel two-fronts bidirectional cross-attention transformer that integrates both wild-type and mutant protein embeddings and their differences, extracted using the ESM2 language model.
4. Thermodynamic compliance is enforced by design. Antisymmetry is built into the architecture using a mirrored input mechanism, while transitivity is incorporated via fine-tuning with a dedicated loss function.
5. JanusDDG outperforms previous sequence-only models and even rivals structure-based methods across several benchmark datasets (S669, S461, S96, PTmut-NR), despite using no structural information.
6. On the Stransitive and Ssym datasets, JanusDDG is one of the few models to satisfy both antisymmetry and transitivity, showing that its predictions are not only accurate but also physically grounded.
7. The model generalizes well to multi-point mutations, handling complex interactions such as epistasis, which often confound traditional approaches.
8. Performance remains robust across a range of mutation distances—both spatial and sequential—indicating its capacity to model local and global mutational effects.
9. In classification tasks, JanusDDG effectively identifies stabilizing mutations, achieving strong metrics across recall, F1 score, and MCC, crucial for protein engineering applications.
10. The core innovation lies in the bidirectional multi-head cross-attention mechanism, which allows mutual context exchange between the wild-type and mutant embeddings, enhancing interpretability and predictive power.
11. JanusDDG represents a step forward in stability prediction by aligning deep learning outputs with physical laws, potentially accelerating protein engineering and variant effect interpretation.
💻Code: github.com/BioComputingUP/Ja…
📜Paper: arxiv.org/abs/2504.03278
#ProteinStability #MutationEffects #DeepLearning #ProteinEngineering #Bioinformatics #Thermodynamics #SequenceOnly #ICLR2025 #ProteinLanguageModels
1
15
1,639
7 Apr 2025
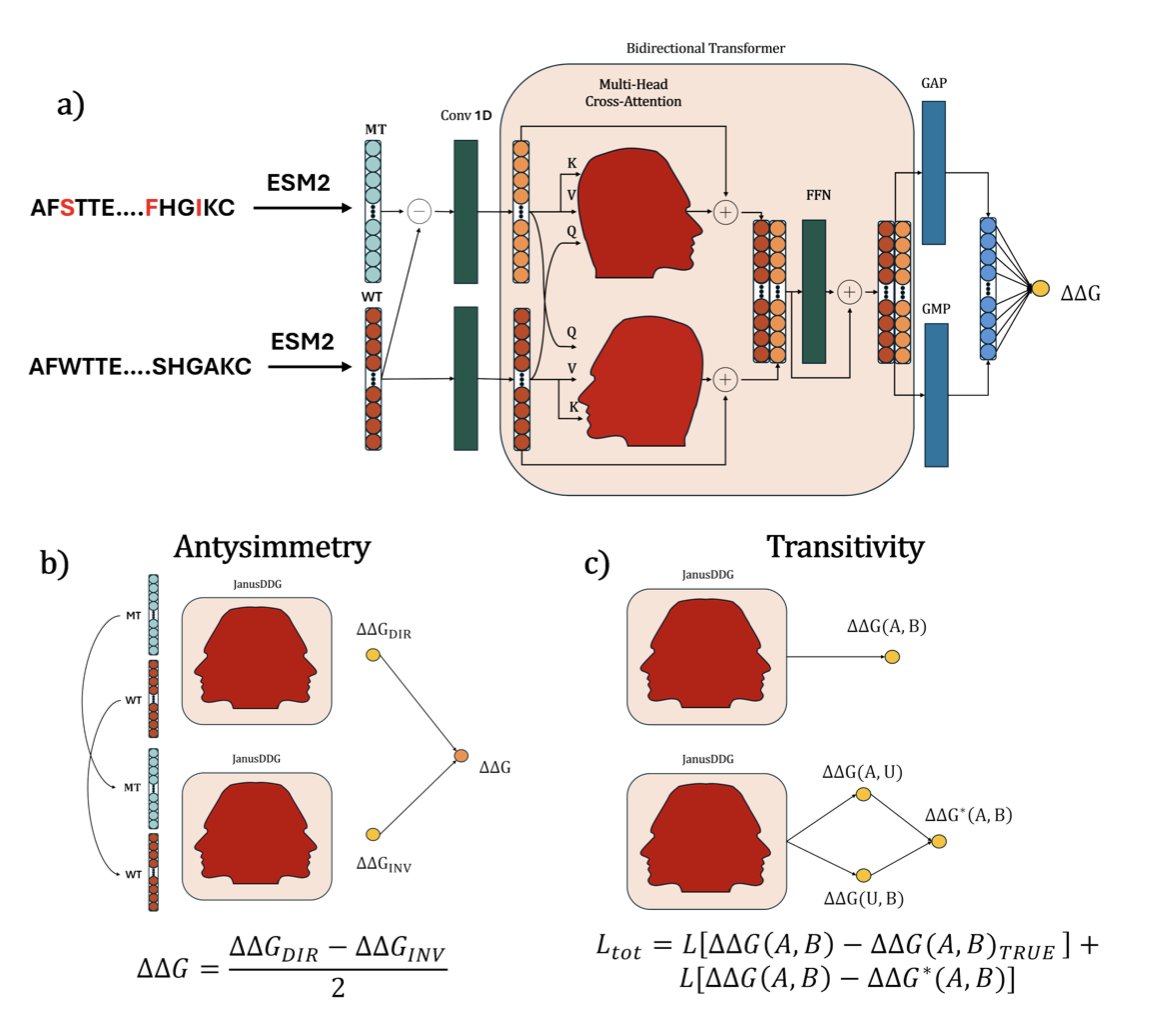
JanusDDG: A Thermodynamics-Compliant Model for Sequence-Based Protein Stability via Two-Fronts Multi-Head Attention
1. JanusDDG is a new sequence-based model that predicts changes in protein stability (ΔΔG) upon mutation, while strictly adhering to thermodynamic principles such as antisymmetry and transitivity.
2. Unlike conventional models that rely on 3D structures, JanusDDG predicts ΔΔG directly from amino acid sequences, making it broadly applicable and computationally efficient.
3. The model introduces a novel two-fronts bidirectional cross-attention transformer that integrates both wild-type and mutant protein embeddings and their differences, extracted using the ESM2 language model.
4. Thermodynamic compliance is enforced by design. Antisymmetry is built into the architecture using a mirrored input mechanism, while transitivity is incorporated via fine-tuning with a dedicated loss function.
5. JanusDDG outperforms previous sequence-only models and even rivals structure-based methods across several benchmark datasets (S669, S461, S96, PTmut-NR), despite using no structural information.
6. On the Stransitive and Ssym datasets, JanusDDG is one of the few models to satisfy both antisymmetry and transitivity, showing that its predictions are not only accurate but also physically grounded.
7. The model generalizes well to multi-point mutations, handling complex interactions such as epistasis, which often confound traditional approaches.
8. Performance remains robust across a range of mutation distances—both spatial and sequential—indicating its capacity to model local and global mutational effects.
9. In classification tasks, JanusDDG effectively identifies stabilizing mutations, achieving strong metrics across recall, F1 score, and MCC, crucial for protein engineering applications.
10. The core innovation lies in the bidirectional multi-head cross-attention mechanism, which allows mutual context exchange between the wild-type and mutant embeddings, enhancing interpretability and predictive power.
11. JanusDDG represents a step forward in stability prediction by aligning deep learning outputs with physical laws, potentially accelerating protein engineering and variant effect interpretation.
💻Code: github.com/BioComputingUP/Ja…
📜Paper: arxiv.org/abs/2504.03278
#ProteinStability #MutationEffects #DeepLearning #ProteinEngineering #Bioinformatics #Thermodynamics #SequenceOnly #ICLR2025 #ProteinLanguageModels
2
7
43
3,436
15 Mar 2025
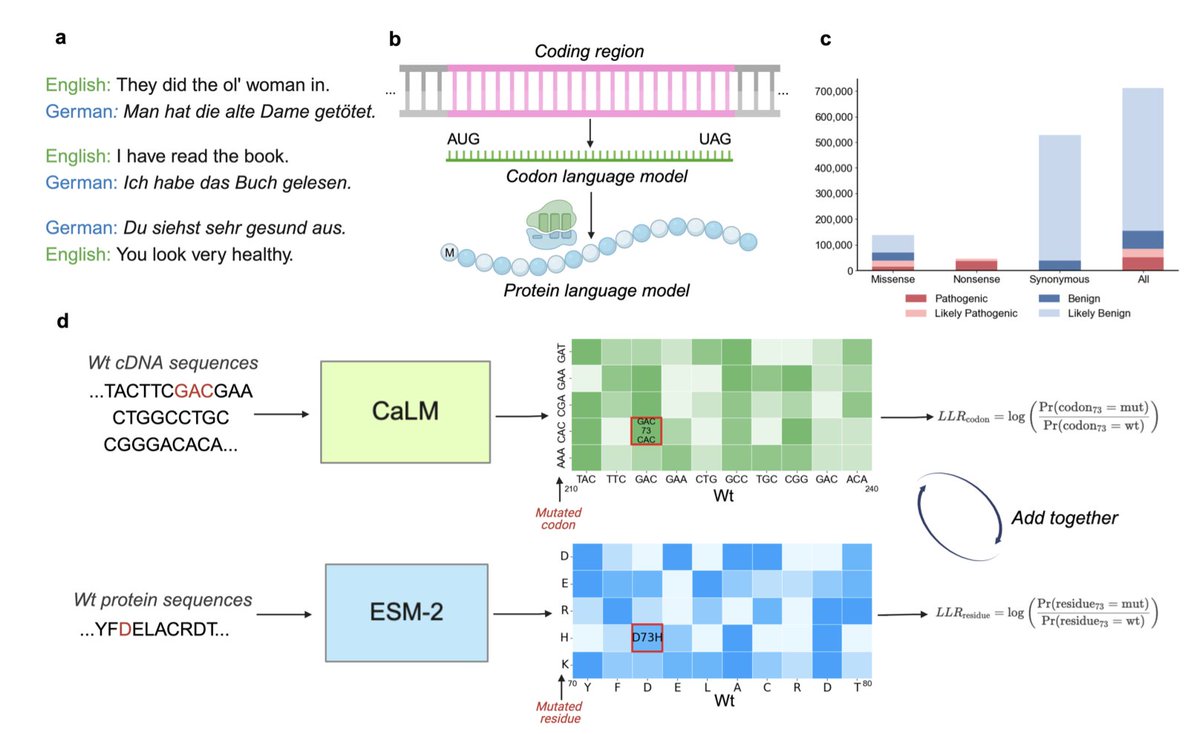
Multilingual Model Improves Zero-Shot Prediction of Disease Effects on Proteins
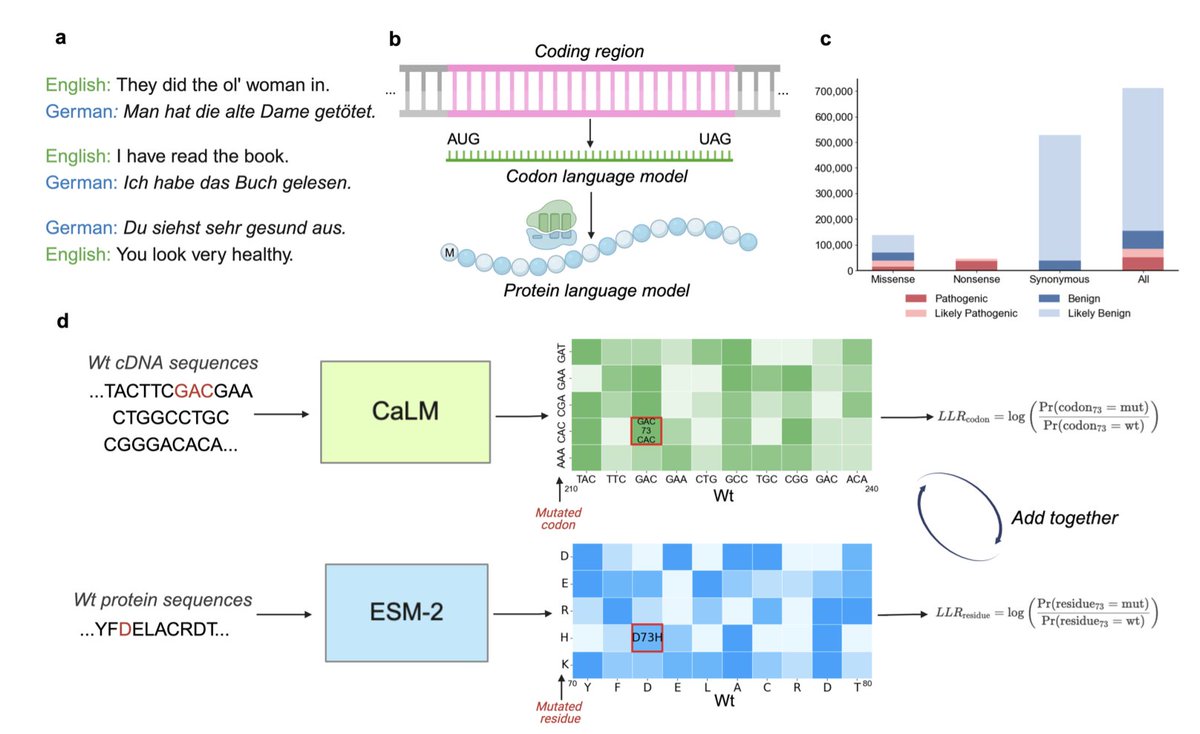
1. The paper introduces a novel approach that combines a codon language model (CaLM) with a protein language model (ESM-2) to enhance the prediction of disease effects caused by mutations in proteins.
2. Unlike traditional single-sequence-based models, this dual representation captures contextual dependencies at both the genetic (codon-level) and protein (amino acid-level) scales, improving classification accuracy for pathogenic and benign mutations.
3. The hybrid model achieves a 3 percent increase in ROC-AUC when predicting disease effects on 137,350 ClinVar missense variants across 13,791 genes, outperforming standalone ESM-2 and CaLM models.
4. By integrating codon and protein sequence information, the model provides a linguistic analogy to multilingual learning, where combining information from different biological scales improves predictive power.
5. The study demonstrates that the codon language model uniquely distinguishes synonymous from nonsense mutations, a capability that protein language models lack, improving overall classification robustness.
6. The model effectively identifies evolutionary constraints on amino acid substitutions, showing that certain codon transitions significantly impact disease outcomes, particularly when shifting between high- and low-codon usage amino acids.
7. Comparative analysis reveals that ESM-2 outperforms CaLM for some pathogenic variants, while CaLM is better at detecting certain synonymous and nonsense mutations, highlighting the complementary strengths of both models.
8. Bayesian optimization was employed to fine-tune the integration of codon and protein effect scores, ensuring optimal weighting for disease variant classification.
9. The hybrid model's insights extend beyond disease classification, offering potential applications in protein engineering, fitness landscape modeling, and genomics-driven precision medicine.
10. Future work will focus on expanding the model to include structural information, refining weighting strategies, and integrating experimental validation to further enhance its predictive accuracy.
💻Code: github.com/Cassie818/Viral-m…
📜Paper: biorxiv.org/content/10.1101/…
#MutationEffects #ProteinModeling #MachineLearning #AIforScience #ComputationalBiology
5
21
2,028
15 Mar 2025
Multilingual Model Improves Zero-Shot Prediction of Disease Effects on Proteins
1. The paper introduces a novel approach that combines a codon language model (CaLM) with a protein language model (ESM-2) to enhance the prediction of disease effects caused by mutations in proteins.
2. Unlike traditional single-sequence-based models, this dual representation captures contextual dependencies at both the genetic (codon-level) and protein (amino acid-level) scales, improving classification accuracy for pathogenic and benign mutations.
3. The hybrid model achieves a 3 percent increase in ROC-AUC when predicting disease effects on 137,350 ClinVar missense variants across 13,791 genes, outperforming standalone ESM-2 and CaLM models.
4. By integrating codon and protein sequence information, the model provides a linguistic analogy to multilingual learning, where combining information from different biological scales improves predictive power.
5. The study demonstrates that the codon language model uniquely distinguishes synonymous from nonsense mutations, a capability that protein language models lack, improving overall classification robustness.
6. The model effectively identifies evolutionary constraints on amino acid substitutions, showing that certain codon transitions significantly impact disease outcomes, particularly when shifting between high- and low-codon usage amino acids.
7. Comparative analysis reveals that ESM-2 outperforms CaLM for some pathogenic variants, while CaLM is better at detecting certain synonymous and nonsense mutations, highlighting the complementary strengths of both models.
8. Bayesian optimization was employed to fine-tune the integration of codon and protein effect scores, ensuring optimal weighting for disease variant classification.
9. The hybrid model's insights extend beyond disease classification, offering potential applications in protein engineering, fitness landscape modeling, and genomics-driven precision medicine.
10. Future work will focus on expanding the model to include structural information, refining weighting strategies, and integrating experimental validation to further enhance its predictive accuracy.
@mikaelboden @n_palpant @ruyic818
💻Code: github.com/Cassie818/Viral-m…
📜Paper: biorxiv.org/content/10.1101/…
#MutationEffects #ProteinModeling #MachineLearning #AIforScience #ComputationalBiology
2
1,254
14 Mar 2025
VenusMutHub: A Systematic Evaluation of Protein Mutation Effect Predictors on Small-Scale Experimental Data
1. The paper introduces VenusMutHub, a comprehensive benchmark designed to evaluate computational models for predicting the effects of protein mutations using small-scale experimental datasets.
2. Unlike previous benchmarks that rely on deep mutational scanning (DMS) data with surrogate readouts, VenusMutHub compiles 905 curated experimental datasets spanning 527 proteins with direct biochemical measurements.
3. The benchmark evaluates mutation effect predictors across key protein properties, including stability, catalytic activity, binding affinity, and selectivity, providing a more realistic assessment for protein engineering applications.
4. The study systematically assesses 23 computational models, categorized into sequence-based, structure-aware, and evolutionary-informed approaches, identifying strengths and limitations in different prediction paradigms.
5. Structure-aware models, particularly inverse folding models like MIF and ESM-IF1, perform best in stability predictions, highlighting the importance of structural information for accurate ∆∆G and ∆Tm estimation.
6. Evolutionary-informed models, such as GEMME and PoET, excel in catalytic activity predictions, suggesting that sequence conservation and evolutionary constraints play a crucial role in enzyme function.
7. Protein-protein interaction (PPI) predictions show significant improvement when using multichain models like ESM-IF1 and ProteinMPNN, demonstrating the necessity of incorporating partner structural information.
8. Selectivity predictions emerge as the most challenging category, with all models performing poorly, suggesting the need for novel approaches that explicitly consider stereochemistry and ligand-binding interactions.
9. Analysis of dataset size impact reveals a critical threshold of 8–13 mutations per dataset, where model performance improves significantly, emphasizing the importance of adequate experimental data for reliable predictions.
10. Future directions include integrating molecular dynamics simulations, enhancing epistatic interaction modeling, and developing uncertainty quantification techniques to improve the reliability of mutation effect predictions.
💻Code: huggingface.co/datasets/AI4P…
📜Paper: arxiv.org/abs/2503.04851
#ProteinEngineering #MutationEffects #ComputationalBiology #Bioinformatics #MachineLearning
4
7
1,087
14 Mar 2025
VenusMutHub: A Systematic Evaluation of Protein Mutation Effect Predictors on Small-Scale Experimental Data
1. The paper introduces VenusMutHub, a comprehensive benchmark designed to evaluate computational models for predicting the effects of protein mutations using small-scale experimental datasets.
2. Unlike previous benchmarks that rely on deep mutational scanning (DMS) data with surrogate readouts, VenusMutHub compiles 905 curated experimental datasets spanning 527 proteins with direct biochemical measurements.
3. The benchmark evaluates mutation effect predictors across key protein properties, including stability, catalytic activity, binding affinity, and selectivity, providing a more realistic assessment for protein engineering applications.
4. The study systematically assesses 23 computational models, categorized into sequence-based, structure-aware, and evolutionary-informed approaches, identifying strengths and limitations in different prediction paradigms.
5. Structure-aware models, particularly inverse folding models like MIF and ESM-IF1, perform best in stability predictions, highlighting the importance of structural information for accurate ∆∆G and ∆Tm estimation.
6. Evolutionary-informed models, such as GEMME and PoET, excel in catalytic activity predictions, suggesting that sequence conservation and evolutionary constraints play a crucial role in enzyme function.
7. Protein-protein interaction (PPI) predictions show significant improvement when using multichain models like ESM-IF1 and ProteinMPNN, demonstrating the necessity of incorporating partner structural information.
8. Selectivity predictions emerge as the most challenging category, with all models performing poorly, suggesting the need for novel approaches that explicitly consider stereochemistry and ligand-binding interactions.
9. Analysis of dataset size impact reveals a critical threshold of 8–13 mutations per dataset, where model performance improves significantly, emphasizing the importance of adequate experimental data for reliable predictions.
10. Future directions include integrating molecular dynamics simulations, enhancing epistatic interaction modeling, and developing uncertainty quantification techniques to improve the reliability of mutation effect predictions.
💻Code: huggingface.co/datasets/AI4P…
📜Paper: arxiv.org/abs/2503.04851
#ProteinEngineering #MutationEffects #ComputationalBiology #Bioinformatics #MachineLearning
552