
New on arXiv | Viral Proteins Reveal Geometry of Protein Language Models (ICML 2026 Workshop)
Using viral proteins as a lens to understand the geometric structure of protein language model embeddings - revealing how PLMs represent evolutionary & functional diversity.
arxiv.org/abs/2606.12609
#AIxBiology #ProteinLanguageModel #ICML2026 #Virology
10
DPLM: Dynamics-aware Protein Language Model via Contrastive Learning Between Sequence and Molecular Dynamics Simulation Trajectory
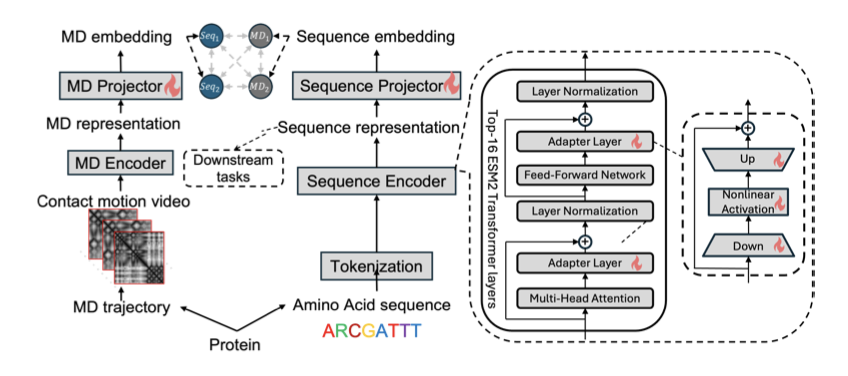
1. Introducing DPLM, a novel protein language model that captures protein dynamics by aligning sequence embeddings with molecular dynamics (MD) trajectory embeddings through contrastive learning, addressing a critical gap in static PLMs.
2. The key innovation lies in using a pretrained Video Vision Transformer (ViViT) to encode MD trajectories as "contact map motion videos," enabling direct learning of spatiotemporal dynamic features without relying on hand-engineered labels or generated conformations.
3. DPLM achieves state-of-the-art performance in zero-shot mutation effect prediction, outperforming ESM2 on 8 out of 9 deep mutational scanning datasets, with particularly strong gains for proteins whose functions depend on conformational flexibility.
4. When adapted with lightweight task-specific heads, DPLM reaches top-tier performance on protein stability prediction (S669 benchmark) and intrinsic disordered region identification (CAID2/CAID3), demonstrating broad transferability.
5. The model employs adapter tuning on ESM2, keeping the base model frozen while adding MD-aware modules in the top 16 layers, ensuring parameter efficiency and preventing catastrophic forgetting of sequence information.
6. Residue-level analysis reveals DPLM embeddings strongly correlate with root-mean-square fluctuation (RMSF), while protein-level clustering outperforms both sequence-only and structure-aware baselines on CATH, deaminase, and kinase benchmarks.
7. A single DPLM-based IDR predictor achieves the best accuracy-efficiency trade-off among CAID competitors, being approximately 10-10,000x faster than structure-dependent methods like SPOT-Disorder2 and AlphaFold-rsa.
8. The work establishes that MD-derived dynamic information, learned through unsupervised cross-modal alignment, yields more biologically meaningful representations than static sequence or structure alone.
💻Code: github.com/UKDPLM/DPLM
📜Paper: biorxiv.org/content/10.64898…
#ProteinLanguageModel #MolecularDynamics #ContrastiveLearning #ProteinDynamics #DeepLearning #Bioinformatics #ESM2 #ViViT #ProteinStability #IntrinsicallyDisorderedProteins #ZeroShotLearning #ComputationalBiology
9
35
3,295
Visualize, explore, and select: A protein language model‑based approach enabling navigation of protein sequence space for enzyme discovery and mining
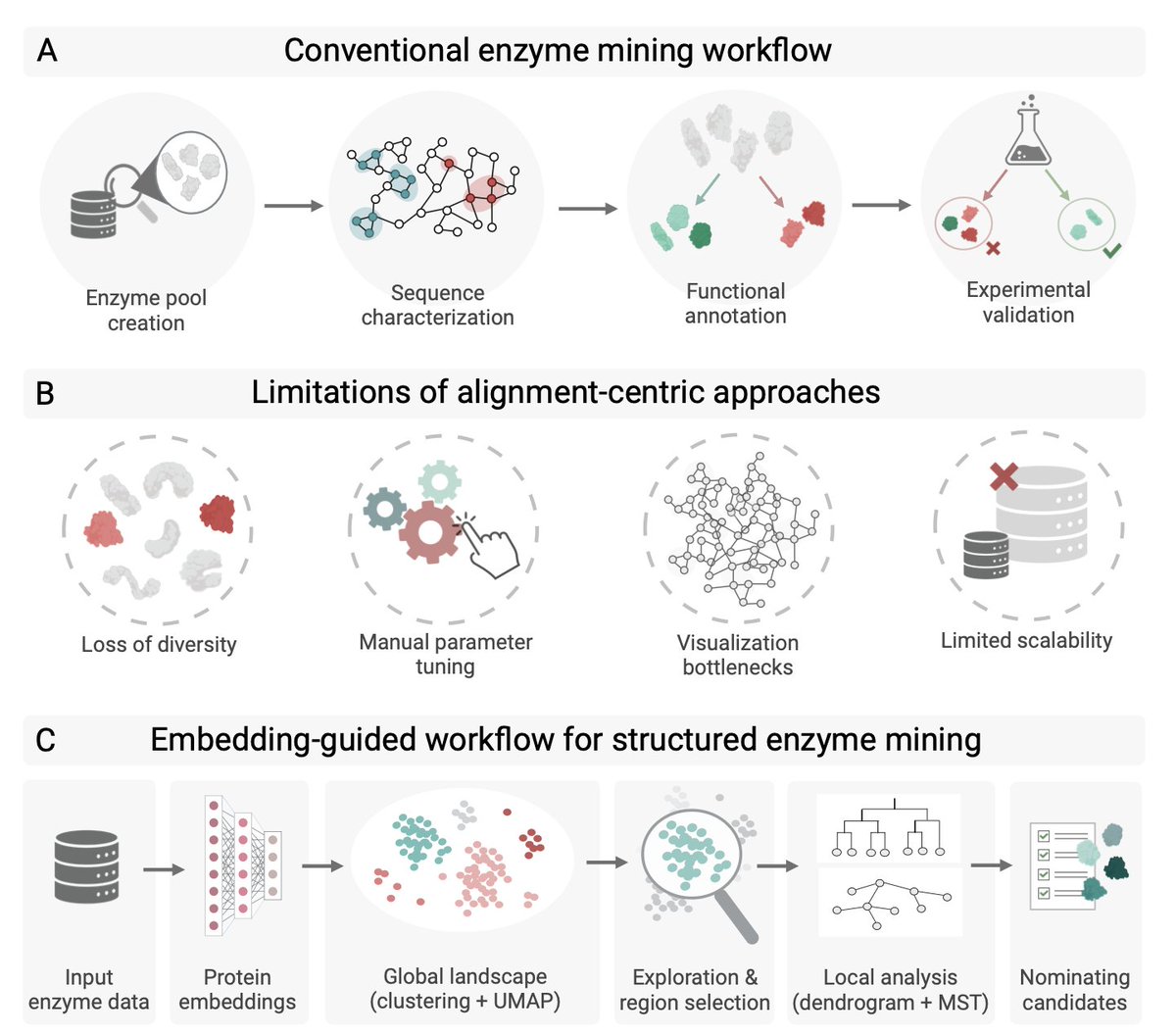
1 This study introduces a novel, alignment‑free workflow that treats enzyme mining as a navigation problem in a high‑dimensional representation space generated by protein language models (pLMs). By moving away from rigid sequence‑identity thresholds, the approach preserves subtle functional relationships that would otherwise be lost in traditional clustering or sequence‑similarity networks.
2 The core pipeline couples pLM embeddings with density‑based clustering (HDBSCAN), dimensionality reduction (UMAP) for global landscape visualization, and a minimum‑spanning‑tree reconstruction to restore connectivity across the embedding manifold. A dendrogram layer then quantifies hierarchical relationships, enabling multi‑scale interpretation from global context to local neighborhoods.
3 In a fully unsupervised test on 5,500 LOV‑domain proteins, the embedding space spontaneously organized sequences by functional effector type and cluster, achieving high k‑nearest‑neighbor agreement for functional labels while showing weak taxonomic coherence. This demonstrates that the latent representations capture biologically relevant signals without any domain‑specific supervision.
4 Applying the method to a heterogeneous PET‑hydrolyzing enzyme space of over 100,000 sequences, researchers anchored the search with experimentally validated PET‑active and PET‑inactive proteins. The embedding‑guided exploration highlighted archaeal, thermophilic candidates proximal to positive anchors, directly addressing industrial constraints such as temperature and pH tolerance.
5 Connectivity‑aware refinement using a minimum‑spanning‑tree and hierarchical distances clarified ambiguous regions where 2D projections suggested divergent clusters. This layer distinguishes closely related variants from structurally distinct neighbors without imposing arbitrary similarity cutoffs, refining candidate nomination.
6 Structural comparisons of seven PET‑proximal candidates revealed that embedding proximity aligns with fold conservation even when sequence identities fall below 30 % (the twilight zone). RMSD analyses showed gradual structural divergence across the embedding continuum, confirming that the representation captures higher‑order structural constraints.
7 The entire workflow is packaged in the open‑source platform SelectZyme, providing interactive visualizations and reproducible pipelines. It scales to more than 100 k sequences, enabling rapid exploration and candidate selection in sparsely annotated sequence landscapes.
8 Flexibility is built into the design: users can filter by organism, predicted properties, or experimental anchors, and can switch between novelty search, optimization around known homologs, or constraint‑aware screening—all within the same embedding framework.
9 The authors note practical caveats such as the dependence on the initial sequence pool, the choice of pLM architecture, and the need for complementary structural or functional assays to validate embedding‑guided predictions. These considerations guide responsible application of the method in real‑world discovery projects.
10 Future directions include integrating activity or stability prediction models, coupling with active‑learning loops, and extending the framework to other enzyme families and multi‑domain architectures, thereby tightening the loop between computation and experiment.
💻Code: github.com/ipb-halle/SelectZ…
📜Paper: biorxiv.org/content/10.64898…
#ProteinEngineering #EnzymeDiscovery #ProteinLanguageModel #MLforBiology #Bioinformatics #StructuralBioinformatics #ComputationalBiology
4
31
2,492
A Contrastive Learning Framework for Efficient Viral Escape Prediction
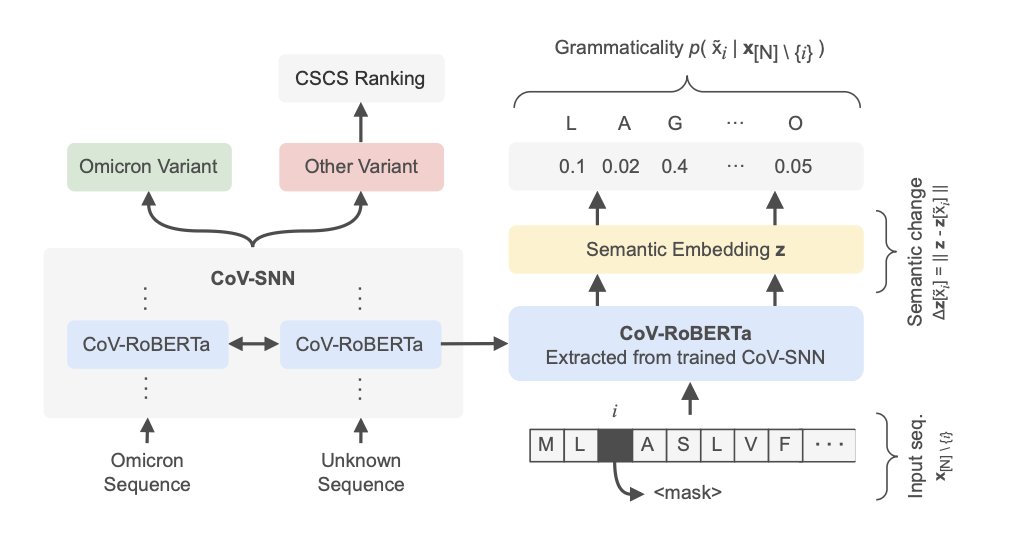
1 The paper introduces CoV‑SNN, a Siamese contrastive learning framework that simultaneously classifies SARS‑CoV‑2 variants and ranks escape‑enhancing mutations, all without any deep‑mutational‑scanning data.
2 A bespoke dataset of 326 K high‑coverage Spike sequences was curated, balancing five variants of concern and ensuring chronological consistency, which provides a clean training ground for the model.
3 CoV‑SNN leverages a lightweight protein language model, CoV‑RoBERTa, and a unique pair‑selection algorithm that builds informative positive/negative pairs on the fly, dramatically reducing over‑fitting on highly similar viral sequences.
4 The escape‑scoring pipeline extends the Constrained Semantic Change Search (CSCS) by mapping semantic shift to embedding distance and grammaticality to an average log‑likelihood, enabling precise prioritization of immune‑evading mutations.
5 Empirical results are striking: 98.8 % multi‑class variant accuracy, 0.909 AUC for zero‑shot classification, and 97.7 % precision at the top 10 % of ranked escape candidates.
6 On wet‑lab‑verified escape datasets, the model attains 0.864 AUC, surpassing all baseline BiLSTM and fitness‑based approaches while executing 125× faster and using 20× less GPU memory.
7 Architectural efficiency is achieved with a 10 K‑token vocabulary and only 52 M parameters, yielding 40× faster inference and 12× lower peak GPU usage compared to larger transformer baselines.
8 Layer‑wise attribution analysis shows lower layers encode semantic change (antigenic variation) while the final layer captures grammaticality (viral fitness), illustrating how the model learns evolutionary constraints.
9 Future directions include extending CoV‑SNN to other viral glycoproteins, handling insertions/deletions, and refining generative prompts for novel variant design.
💻Code: github.com/smtnkc/CoV-SNN
📜Paper: doi.org/10.1109/TCBBIO.2026.…
#ComputationalBiology #ProteinLanguageModel #ViralEvolution #Bioinformatics #MachineLearning #SARSCoV2 #ContrastiveLearning #DeepLearning #AntibodyEscape
3
12
1,123
AbAffinity: A Large Language Model for Predicting Antibody Binding Affinity against SARS-CoV-2
1 AbAffinity achieves state-of-the-art performance in predicting antibody binding affinity against SARS-CoV-2, with Pearson correlation of 0.655 and Spearman correlation of 0.608, significantly outperforming existing methods including DG-Affinity, ESM-2, and AbLang.
2 The model is built on a fine-tuned ESM-2 architecture with 33 layers and 650M parameters, specifically trained on 71,834 unique scFv antibody sequences with experimentally measured binding affinities against a conserved HR2 peptide region of SARS-CoV-2.
3 Unlike generic protein language models, AbAffinity learns SARS-CoV-2-specific binding patterns through task-specific fine-tuning, enabling it to capture mutation effects and create meaningful embedding spaces that correlate smoothly with binding affinity.
4 The model generates interpretable residue-residue attention maps that highlight complementarity-determining regions (CDRs) as critical for binding prediction, providing biological insights into strong versus weak binding antibodies.
5 AbAffinity's embeddings demonstrate unexpected versatility by capturing thermostability properties of antibodies without explicit training on this attribute, and enable superior performance on downstream classification tasks compared to baseline embeddings.
6 The model is publicly available through PyPI, allowing seamless integration into antibody design workflows for therapeutic development against COVID-19 and potentially other infectious diseases.
💻Code: pypi.org/project/AbAffinity/
📜Paper: arxiv.org/abs/2603.04480
#AbAffinity #AntibodyDesign #SARSCoV2 #BindingAffinity #ProteinLanguageModel #ComputationalBiology #AIforScience #TherapeuticAntibodies

6
22
1,785
DecoderTCR: Compositional Pretraining and Entropy-Guided Decoding for TCR-pMHC Interactions
1 The authors introduce DecoderTCR, a masked language model framework that achieves 0.96 AUROC for zero-shot pMHC binding prediction and 0.76 AUROC for epitope-specific TCR recognition, approaching supervised baselines without epitope-specific training.
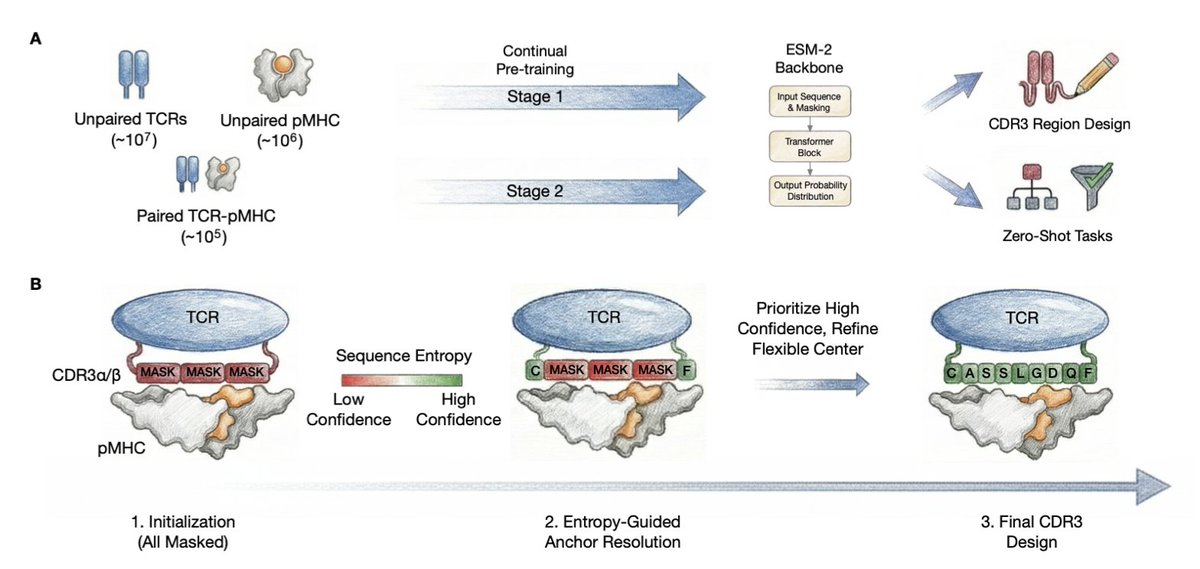
2 The key innovation is a two-stage continual pre-training curriculum that bridges the 100x data gap between abundant unpaired sequences (~10^7 TCRs, ~10^6 pMHCs) and sparse paired interactions (~10^5) through component-specific masking strategies.
3 Stage 1 learns component representations from marginal data using functionally-informed masking, while Stage 2 refines cross-chain dependencies from limited paired data with joint masking of CDR and peptide positions, providing implicit experience replay that prevents catastrophic forgetting.
4 The second major contribution is Iterative Entropy-Guided Refinement (IEGR), a non-autoregressive decoding algorithm that resolves high-confidence anchor positions first before refining high-entropy flexible regions, respecting the biophysical hierarchy of CDR3 loops.
5 Learned representations recover structural contacts without coordinate supervision, with interaction scores correctly identifying MHC anchor positions at peptide termini and TCR contact positions at the central bulge.
6 Experimental validation reveals a critical prediction-generation gap: while the model discriminates binders from non-binders at 0.76 AUROC, zero-shot generation of functional TCRs remains challenging with only 1 of 20 designed candidates showing binding above background.
7 The work establishes the first rigorous characterization of this prediction-generation gap in TCR design, providing a concrete benchmark for the field and highlighting that strong discrimination does not yet yield reliable generation.
8 The approach demonstrates that compositional pre-training enables sample-efficient learning for multi-component protein interfaces where paired data is scarce but marginal data is abundant, with principles extendable to other biological systems.
💻Code: github.com/czbiohub-chi/Deco…
📜Paper: biorxiv.org/content/10.64898…
#TCR #pMHC #Immunology #ProteinLanguageModel #ComputationalBiology #MachineLearning #TCellReceptor #AntigenRecognition #GenerativeAI #Bioinformatics
4
16
1,146
No Generation Without Representation: Efficient Causal Protein Language Models Enable Zero-Shot Fitness Estimation
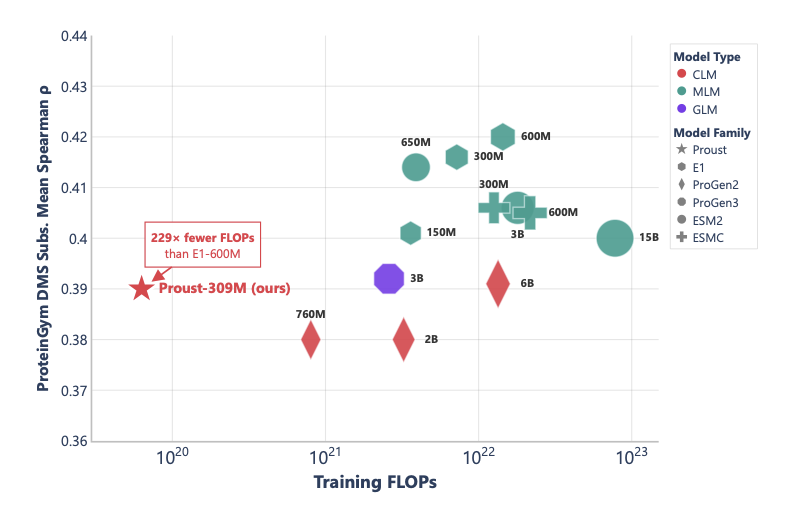
1 Proust, a 309 M-parameter causal protein language model, delivers MLM-grade fitness prediction while still able to generate sequences, closing the long-standing divide between masked and autoregressive PLMs.
2 Trained on 33 B tokens in 40k GPU-hours, it reaches Spearman ρ = 0.39 on ProteinGym substitutions, matching ESM2-650 M within 0.01 yet using 62× fewer FLOPs and E1-600 M within 0.03 at 229× less cost.
3 On 69 indel assays Proust sets a new single-sequence state-of-the-art (ρ = 0.52), outperforming models up to 20× larger and showing that causal scoring naturally handles length-changing mutations.
4 Architectural efficiency—not protein-specific tricks—drives the gains: GQA-S2 attention with shared K/V, cross-layer value residuals, key-offset bigram heads, Canon depthwise causal convolutions, plus Muon optimizer with Newton–Schulz orthogonalization.
5 Internal logit-lens entropy predicts when retrieval helps: std-dev of per-position entropy correlates −0.40 with MSA benefit, offering a cheap heuristic for test-time compute allocation.
6 Attention maps reveal biophysical knowledge without structural input: 65 % of weight lands >20 residues away, hydrophobic residues receive highest attention, and core positions show lowest prediction entropy, mirroring evolutionary constraints.
7 The model suppresses Trp at 41 % and Cys at 22 % of sites, aligns positional uncertainty with N/C-terminal variability, and recognizes functional motifs such as CxxC zinc fingers via lowered entropy.
8 Limitations remain: thermostability prediction still trails structure-aware methods, and retrieval-augmented MLMs outperform when compute budget is unconstrained, leaving room for future scaling.
💻Code: github.com/Furkan9015/proust…
📜Paper: arxiv.org/abs/2602.01845
#proteinlanguageModel #causalLM #fitnessPrediction #zeroShot #efficientAI #computationalBiology
1
3
17
1,670
Minimal-Action Discrete Schrödinger Bridge Matching for Peptide Sequence Design
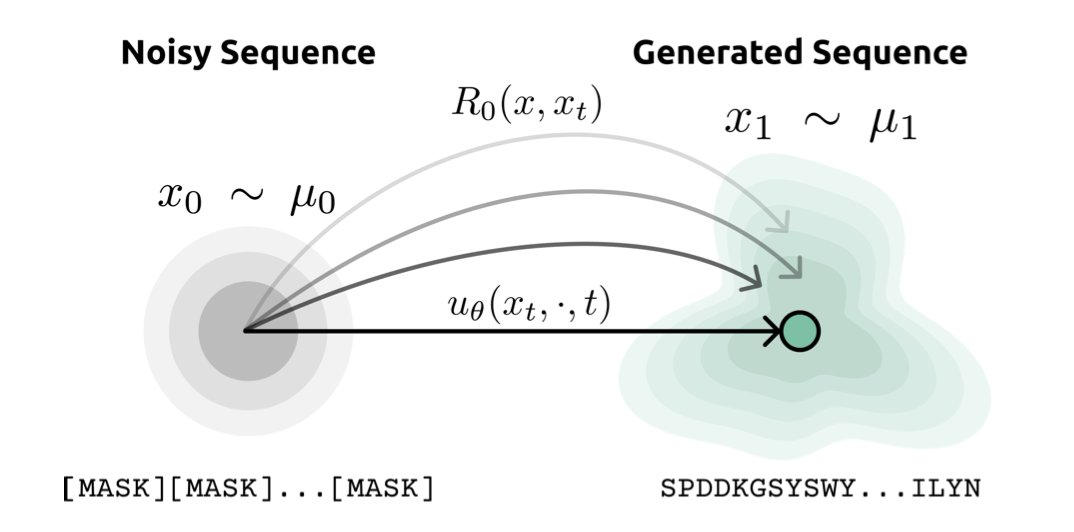
1 MadSBM reframes peptide design as a rate-based transport problem on an amino-acid edit graph, steering generation through high-likelihood neighborhoods instead of forcing it across chemically implausible intermediates.
2 The model couples a frozen ESM-2 language model to supply biologically informed reference dynamics, then learns only a lightweight 50 M-parameter control field that tilts the CTMC rates toward the data distribution.
3 Training collapses to a simple cross-entropy loss on masked tokens; no expensive forward–backward bridge iterations are required, yet the learned control field provably converges to the minimal-action Schrödinger bridge.
4 Generation runs backward in continuous time with a Poisson jump process; only 32 discrete steps yield peptide sequences whose ESM-2 pseudo-perplexity beats a 150 M-parameter discrete diffusion baseline trained on the same data.
5 Objective-guided sampling is introduced for the first time in a discrete Schrödinger bridge: an external binding classifier reranks 16 candidates per jump, raising predicted affinities above known experimental binders for three of four tested targets.
6 Ablations show that time-gating the ESM prior is critical: removing it increases perplexity, while fully dropping ESM forces the DiT to learn biophysics from scratch, validating the hybrid prior-plus-control design.
7 Theoretical guarantees relate cross-entropy minimization to action minimization, bound control error in KL divergence, and prove convergence of the time-discretized sampler as step size → 0.
💻Code: github.com/pranamchatterjee-…
📜Paper: arxiv.org/abs/2601.22408
#peptidedesign #generativemodeling #schrodingerbridge #proteinlanguageModel #optimaltransport
9
41
2,626
Seq2Pocket: Augmenting protein language models for spatially consistent binding site prediction
1 Seq2Pocket directly attacks the “Swiss-cheese” problem of fragmentary pockets that plague current protein language model (pLM) predictors, lifting DCC recall on LIGYSIS by 11 % without inflating false positives.
2 A light-weight, embedding-driven smoothing network (3-layer MLP) learns to fill spatial gaps inside predicted cavities; trained only on CryptoBench apo sites, it generalizes across datasets and adds only 1 % extra residues on average.
3 The authors coin the Pocket Fragmentation Index (PFI): the mean number of predicted clusters that overlap one true pocket. Their Mean-Shift surface clustering keeps PFI ≈ 1.9 while DBSCAN-style fragmentation can exceed 4, exposing why high DCC scores can mislead.
4 Re-annotating sc-PDB to create sc-PDB-enhanced ( 234 % extra pockets including ions) gives the same ESM-2 3B backbone a >15 % jump in pocket-level recall, proving that richer supervision beats architectural tinkering.
5 On the cryptic-site benchmark CryptoBench the smoothed pLM reaches 76.7 % DCCtop-(N 2) recall, clearly outperforming the prior best of 73 % and showing that sequence models can spot druggable conformational states without holo templates.
6 The entire pipeline—from residue logits to ranked 3D pockets—runs in minutes on a single GPU, releasing practical models for both orthosteric and cryptic site discovery in early-stage drug discovery.
💻Code: github.com/skrhakv/seq2pocke…
📜Paper: biorxiv.org/content/10.64898…
#proteinlanguagemodel #bindingsiteprediction #crypticpockets #drugdiscovery #bioinformatics #ESM2

8
36
2,185
Context-Aware Protein Representations Using Protein Language Models and Optimal Transport
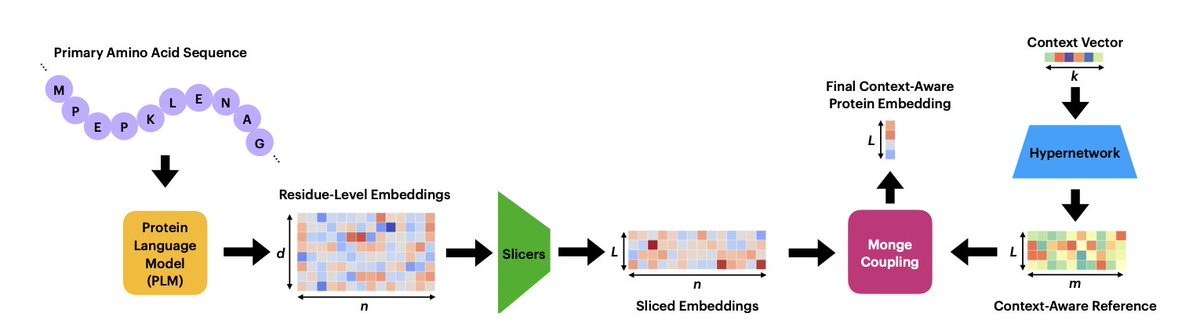
1. COPTER introduces a context-aware pooling mechanism that fuses frozen residue-level protein language model (PLM) embeddings with biological context via sliced-Wasserstein optimal transport, producing a single adaptive protein vector.
2. A lightweight hypernetwork maps any context vector—cell embedding, gene-expression profile, or epitope embedding—into task-specific reference distributions that steer the optimal-transport alignment, letting the same protein sequence yield different representations in different milieus.
3. On 156 cell-type-specific protein networks for rheumatoid-arthritis and inflammatory-bowel-disease target nomination, COPTER reaches near-perfect AP@5 and AUROC, clearly outperforming PINNACLE and GAT baselines without fine-tuning the underlying PLM.
4. t-SNE visualisations show that even random-initialisation COPTER separates proteins by cell context in zero-shot mode, confirming that the transport-based conditioning itself embeds contextual distance.
5. For genetic-perturbation response prediction across Replogle and Norman K562/RPE1 screens, COPTER equals or beats GEARS on single-gene settings, while double-perturbation accuracy drops, hinting that isoform simplification limits signal.
6. TCR–epitope-binding experiments on RN, NA and ET benchmarks give COPTER top accuracy in two of three setups; F1 and AUPRC lag, reflecting dataset imbalance and heuristic negative sampling rather than model failure.
7. The framework is PLM-agnostic: ESM-2 (8 M & 650 M), ProGen2-small and ProGen2-large all plug in frozen, so users can scale model size without retraining the context module.
8. Because only the hypernetwork and final MLP are learnt, COPTER needs modest data and compute, opening the door to rapid context prototyping for single-cell atlas projects or precision-oncology panels.
💻Code: github.com/navidnaderi/COPTE…
📜Paper: biorxiv.org/content/10.64898…
#proteinlanguageModel #optimaltransport #contextawareAI #singlecell #drugtarget #TCR #bioinformatics #computationalbiology
1
7
44
2,751
Protein Language Modeling Beyond Static Folds Reveals Sequence-Encoded Flexibility
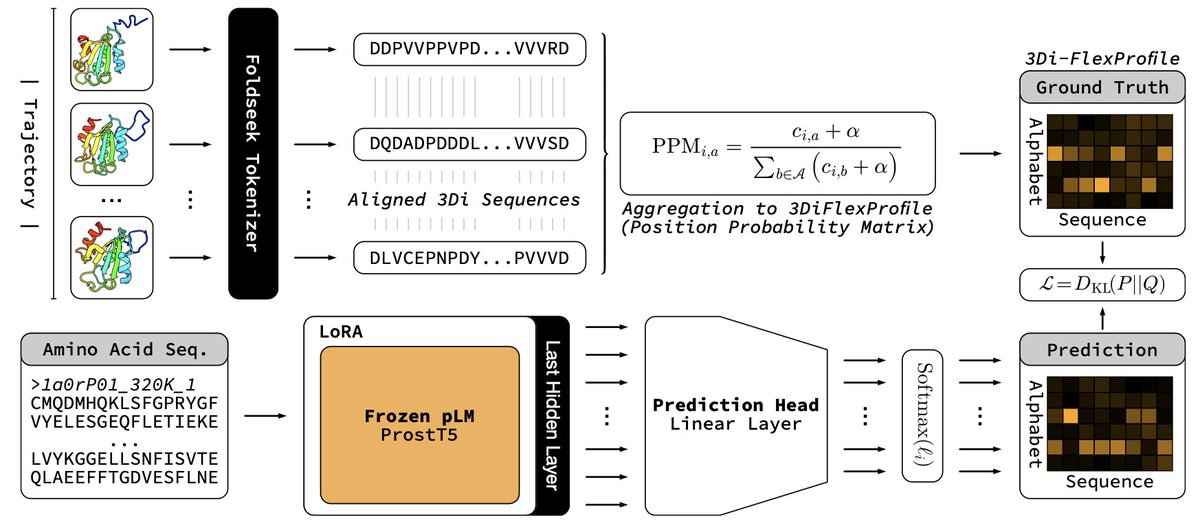
1 ProtProfileMD translates a single amino-acid sequence into a per-residue probability profile over 3Di structural tokens, capturing how often each local conformation is visited during MD without running any simulation at query time.
2 The model distills 5 398 CATH domains’ 320 K trajectories into a 3Di “FlexProfile”; entropy of the predicted profile correlates with RMSF at PCC 0.60, flagging flexible or disordered segments genome-wide in seconds.
3 Supervised LoRA fine-tuning of ProstT5 (only 2.2 M trainable params) lets the pLM learn an implicit energy-weighted ensemble, bypassing costly all-atom ensemble generation while staying proteome-scalable (~0.1 s/protein).
4 Remote homology detection benchmarked on SCOPe shows ROC-AUC gains at Family, Superfamily and Fold levels over standard 3Di search, proving that dynamics fingerprints boost sensitivity beyond static structure tokens.
5 Framework is alphabet-agnostic: any structural vocabulary (PB, DSSP, internal coords) or experimental ensemble (cryo-EM, NMR) can replace 3Di, making FlexProfiles a generic route to encode motion inside language models.
💻Code: github.com/finnlueth/ProtPro…
📜Paper: biorxiv.org/content/10.64898…
#proteindynamics #proteinlanguageModel #computationalBiology #bioinformatics #mdSimulation #structuralAlphabet #homologyDetection
6
31
2,023
Sequence-Only Prediction of Antibody Fab Thermostability Using Protein Language Model Embeddings
1. This study introduces a novel approach to predict the thermostability of antigen-binding fragments (Fabs) using embeddings from IgBERT, a protein language model fine-tuned on antibody sequences. This method offers a scalable and cost-effective alternative to traditional experimental approaches for assessing antibody developability.
2. The core innovation lies in leveraging machine learning to directly predict Fab melting temperatures (Tm) from sequence data alone. The model achieved a Pearson correlation coefficient of 0.77 on an internal test set, demonstrating its potential for rapid and accurate differentiation of stable and unstable Fabs.
3. The study highlights the limitations of traditional statistical models, which often fail to generalize beyond specific datasets. In contrast, the ML model trained on IgBERT embeddings outperformed baseline methods, even when applied to a diverse set of clinical-stage therapeutics.
4. An interesting finding is that the model's generalizability is influenced by the distributional similarity of embeddings rather than sequence identity. This suggests that contextual information captured by protein language models can be more informative for biophysical predictions than raw sequence data.
5. The authors also explored the impact of embedding filtering and dimensionality reduction techniques, such as t-SNE, to improve model performance and interpretability. These methods helped enhance the correlation between predicted and actual Tm values.
📜Paper: biorxiv.org/content/10.64898…
#AntibodyThermostability #ProteinLanguageModel #MachineLearning #Bioinformatics #AntibodyEngineering

2
10
1,241

22 Dec 2025
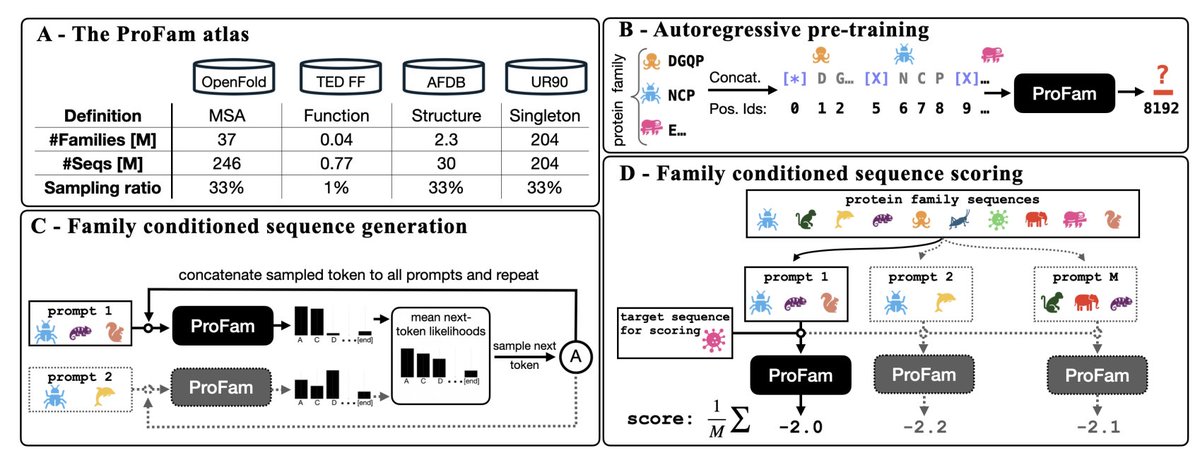
ProFam: Open-Source Protein Family Language Modelling for Fitness Prediction and Design
1. ProFam-1 is a 251 M-parameter autoregressive protein-family language model (pfLM) that reaches state-of-the-art zero-shot fitness prediction on ProteinGym: Spearman ρ = 0.47 for substitutions and 0.53 for indels.
2. Instead of masking or diffusion, ProFam-1 is trained with next-token prediction on unaligned, concatenated homologous sequences, which naturally scores insertions, deletions and arbitrary-length variants under one unified likelihood.
3. The training corpus, ProFam Atlas, bundles ~40 M protein families (≈ 481 M sequences) drawn from AlphaFold DB clusters, TED FunFams, OpenFold MSAs and UniRef90 singletons, giving researchers an open, multi-definition family resource.
4. At inference, the model can be prompted with any subset of family members; ensembling over multiple random prompts (test-time scaling) systematically improves both fitness prediction and sequence diversity without extra training.
5. Generated sequences recapitulate natural conservation, covariance and length distributions; synthetic MSAs from single-seed prompts raise ColabFold lDDT from 0.5 (no MSA) to 0.7, narrowing the gap to natural MSAs (0.9).
6. All weights, training and inference code, and the ProFam Atlas are released under permissive licenses, lowering the barrier for community-driven protein design and benchmarking.
💻Code: github.com/alex-hh/profam/
📜Paper: biorxiv.org/content/10.64898…
#ProteinLanguageModel #ProteinDesign #OpenScience #ProteinGym #AlphaFold #SyntheticMSA
1
1
19
1,304
22 Dec 2025
Structure-Aware Antibody Design with Affinity-Optimized Inverse Folding
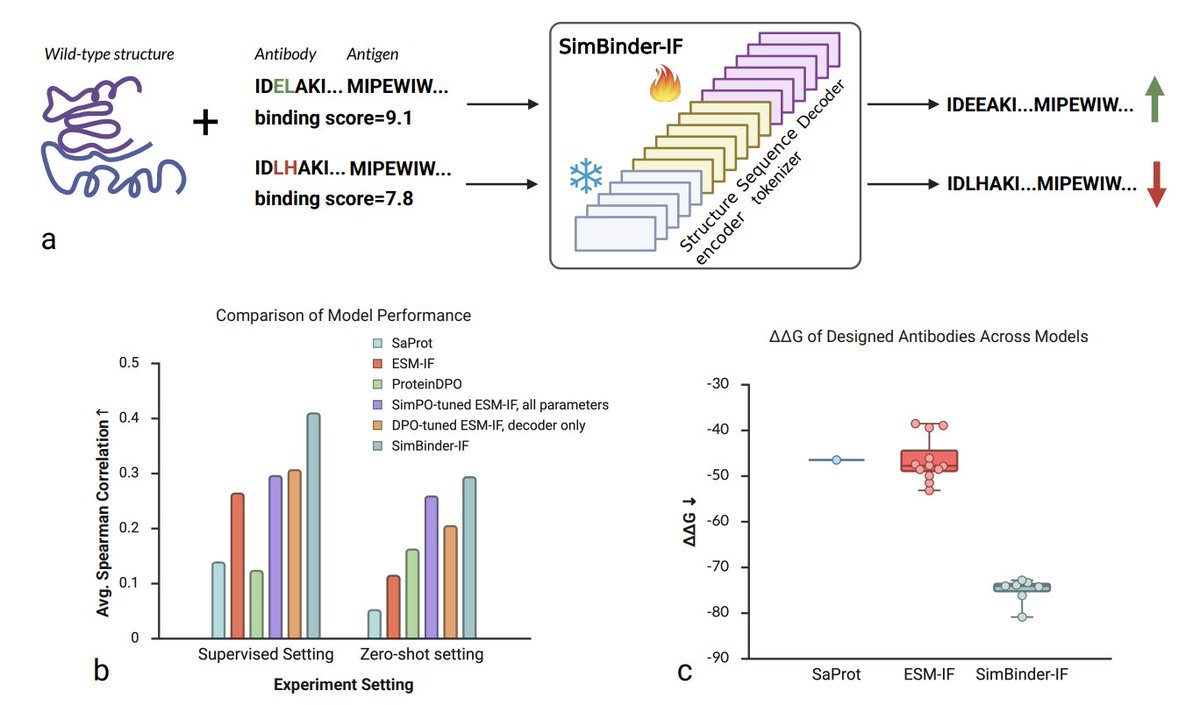
1. SimBinder-IF turns ESM-IF into a high-affinity antibody generator by freezing the structure encoder and tuning only 18 % of the parameters, slashing GPU memory and wall-clock time while boosting performance.
2. Using Simple Preference Optimization (SimPO) instead of DPO removes the need for a reference model, aligning the training objective with the actual inference metric—log-likelihood—and eliminating ~50 % of mis-ranked pairs seen in DPO.
3. On the 11-assay AbBiBench benchmark, SimBinder-IF raises the mean Spearman correlation between predicted log-likelihood and measured binding affinity from 0.264 to 0.410 ( 55 %) in the supervised setting.
4. In zero-shot tests on four unseen antigen–antibody complexes, the correlation jumps from 0.115 to 0.294 ( 156 %), demonstrating strong generalization without additional training data.
5. Top-10 precision for ≥10-fold affinity improvements is markedly higher than vanilla ESM-IF, ProteinDPO, and SaProt, making it the most reliable filter for experimental follow-up.
6. A case study redesigning anti-influenza antibody F045-092 for pdmH1N1 produced variants with a mean FoldX ΔΔG of –75.16 kcal mol⁻¹ versus –46.57 kcal mol⁻¹ from standard ESM-IF, confirming tighter antigen engagement.
7. Structural quality checks (pLDDT, ipLDDT, PTM, epitope SASA, ProteinMPNN likelihood) show that higher affinity does not come at the cost of foldability or interface credibility.
8. Binding-site analysis reveals that SimBinder-IF designs preferentially target the HA head region—the major immunodominant site—suggesting a natural immune-response-like mode of neutralization.
9. The decoder-only update strategy preserves the base model’s structural priors, prevents catastrophic forgetting, and reduces trainable parameters to ~25 M, enabling single-GPU training in under 13 h per epoch.
10. Limitations include a head-centric epitope bias that may limit breadth, single-objective optimization (affinity only), and reliance on in-silico metrics; future work will integrate multi-objective SimPO, selective encoder unfreezing, and high-throughput experimental loops.
📜Paper: arxiv.org/abs/2512.17815v1
#AntibodyDesign #ProteinLanguageModel #InverseFolding #AffinityMaturation #SimPO #ESM #ComputationalBiology #AI4Science
2
16
1,786
19 Dec 2025
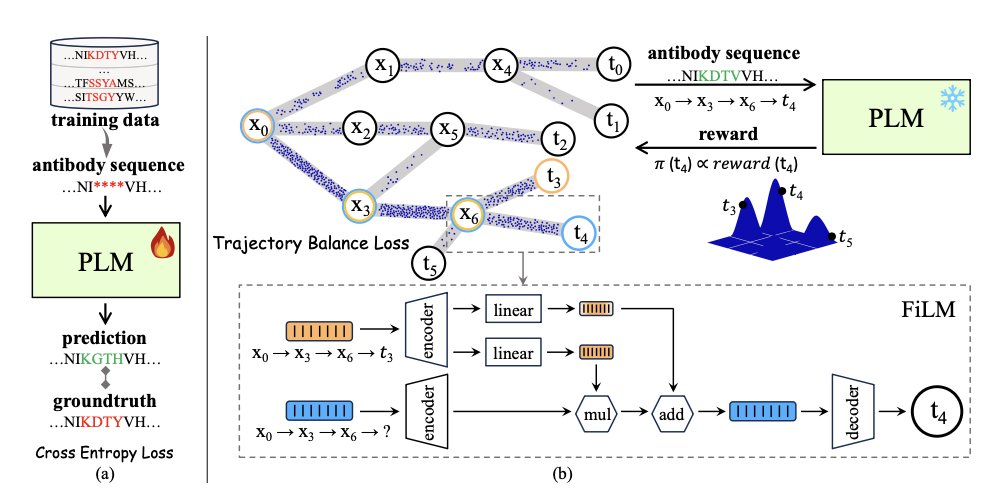
Synergy of GFlowNet and Protein Language Model Makes a Diverse Antibody Designer
1. A new study by Yin et al. introduces PG-AbD, a novel framework combining Generative Flow Networks (GFlowNets) and Protein Language Models (PLMs) to design diverse and potent antibody candidates. This approach significantly enhances the diversity of generated antibodies while maintaining high developability and novelty.
2. The framework leverages a Products of Experts (PoE) model, integrating global constraints from PLMs and local constraints from Potts Models, to guide GFlowNet in exploring the vast antibody sequence space. This joint training paradigm allows for the generation of antibodies with unique binding modes.
3. PG-AbD demonstrates remarkable performance on multiple benchmarks, achieving a 13.5% improvement in diversity on RabDab and a 31.1% improvement on SabDab, outperforming existing methods. This highlights its potential for real-world antibody discovery.
4. The study also includes complex structural binding simulations, showing that generated antibodies form stable and regular 3D structures with antigens. This validates the practical applicability of PG-AbD in designing functional antibodies.
5. PG-AbD operates in a training-data-free mode, making it suitable for both novel antibody design and optimization tasks. This flexibility is crucial for scenarios with limited training data.
📜Paper: ojs.aaai.org/index.php/AAAI/…
#AntibodyDesign #GFlowNet #ProteinLanguageModel #ComputationalBiology #AIinMedicine
4
29
2,651
18 Dec 2025
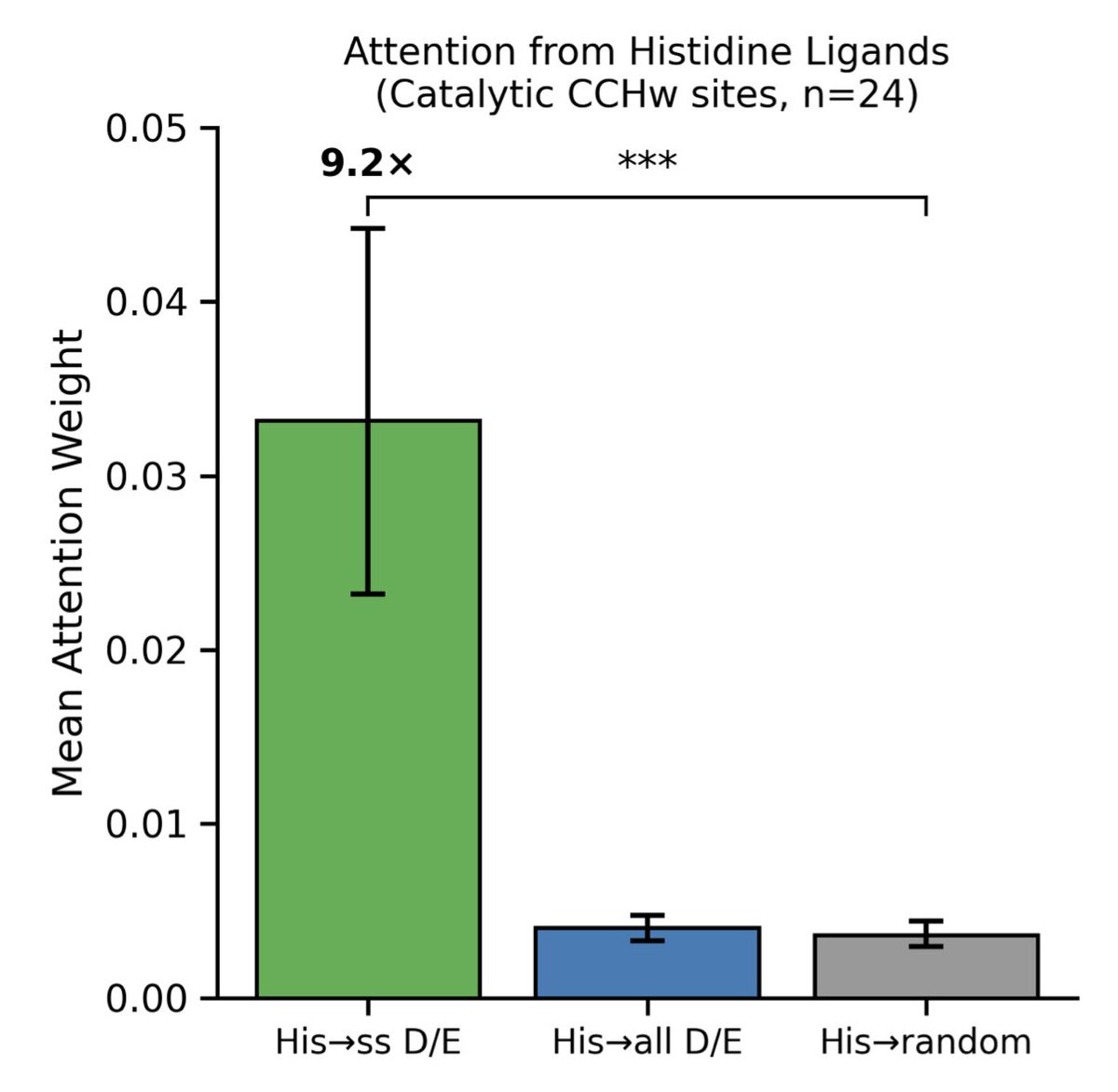
Protein Language Model Embeddings Distinguish Catalytic from Structural Zinc-Binding Sites with Interpretable Attention Signatures
1. A novel study by Karen Sargsyan demonstrates that ESM-2 embeddings can accurately classify catalytic versus structural zinc-binding sites in proteins using sequence information alone. This is significant because these sites share similar coordination geometries, making them difficult to distinguish using traditional methods.
2. The study achieved an impressive ROC-AUC of 0.931 using a random forest classifier on a diverse set of 73 zinc proteins. This significantly outperforms the motif-based baseline (AUC = 0.759), highlighting the power of protein language models in capturing nuanced functional differences.
3. Attention analysis revealed that histidine ligands in catalytic sites attend 9.2-fold more strongly to second-shell carboxylate residues, which are crucial for catalysis. This mechanistic interpretability shows that the model learns biologically meaningful features from evolutionary sequence patterns.
4. The sequence-only approach is particularly valuable for large-scale metalloproteome annotation, especially for the thousands of predicted zinc-binding proteins lacking experimental characterization. It complements structure-based methods and bypasses uncertainties in computationally predicted structures.
5. The study's rigorous evaluation strategy ensures that performance metrics reflect true generalization capacity. Each test protein represents an independent evolutionary lineage, sharing less than 30% sequence identity with training examples. This prevents homology-based information leakage.
6. The findings suggest that protein language models can extract functional signatures directly from evolutionary patterns, making them well-suited for functional annotation of metalloenzymes. Future work could involve integrating larger datasets and exploring other metalloenzyme families.
📜Paper: doi.org/10.26434/chemrxiv-20…
#ProteinLanguageModel #ZincBindingSites #Bioinformatics #ComputationalBiology
1
1
19
1,280
16 Dec 2025
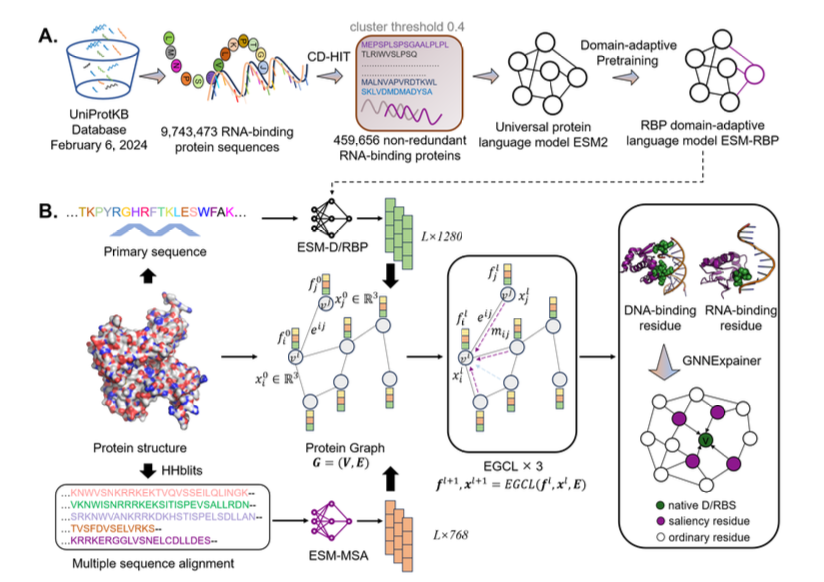
Accurate Nucleic Acid-Binding Residue Identification based Domain-Adaptive Protein Language Model and Explainable Geometric Deep Learning
1. A novel method named GeSite is proposed for predicting nucleic acid-binding residues in proteins, integrating domain-adaptive protein language models and E(3)-equivariant graph neural networks. This approach significantly outperforms existing methods, achieving superior accuracy in identifying both DNA and RNA binding sites.
2. The core innovation lies in the domain-adaptive protein language model, which is specifically fine-tuned on large-scale nucleic acid-binding protein sequences. This allows for more accurate sequence characterization compared to general protein language models, enhancing the prediction performance.
3. GeSite leverages an E(3)-equivariant graph neural network to capture spatial relationships within protein structures. This not only improves prediction accuracy but also provides interpretability by identifying key functional domains involved in nucleic acid binding.
4. The method demonstrates excellent performance across multiple benchmark datasets, with MCC values of 0.522 for DNA-binding residues and 0.326 for RNA-binding residues, representing substantial improvements over previous state-of-the-art methods.
5. Interpretability analysis using GNNExplainer reveals that GeSite effectively identifies nucleic acid-binding domains, even in cases where these domains are spatially distant from the target residues. This highlights the model's ability to capture long-range interactions.
6. The study highlights the potential of combining domain-specific language models with geometric deep learning for advancing our understanding of protein-nucleic acid interactions, with implications for drug development and functional genomics.
💻Code: github.com/pengsl-lab/GeSite
📜Paper: ojs.aaai.org/index.php/AAAI/…
#ProteinNucleicAcidBinding #GeometricDeepLearning #ProteinLanguageModel #Bioinformatics #AIinBiology
1
3
32
2,053
17 Nov 2025
MotifAE Reveals Functional Motifs from Protein Language Model: Unsupervised Discovery and Interpretability Analysis
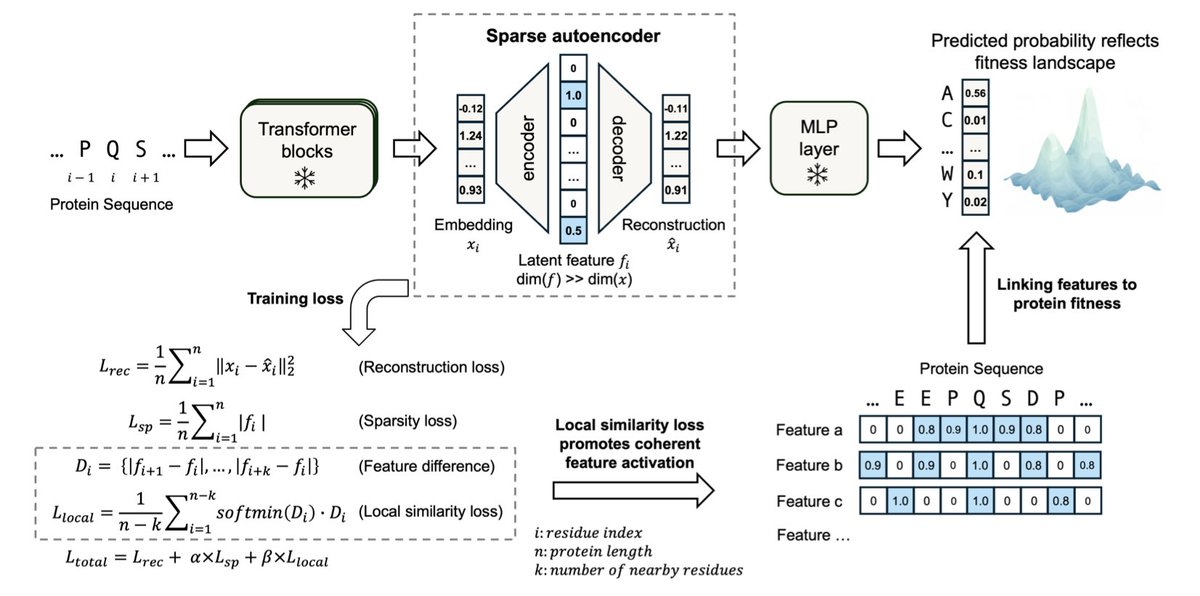
1. MotifAE is an innovative unsupervised framework designed to discover functional motifs from protein language models, specifically leveraging the ESM2 model. This approach captures evolutionary-scale sequence regularities, enabling the identification of motifs that mediate critical biological processes like folding, binding, and catalysis.
2. The core of MotifAE is a sparse autoencoder (SAE) architecture that projects ESM2 embeddings into a sparse latent space. By introducing a local similarity loss, MotifAE encourages coherent latent feature activations, reflecting the sequential nature of protein motifs and improving motif discovery compared to standard SAEs.
3. When benchmarked against known ELM motifs, MotifAE achieves a median AUROC of 0.88, significantly outperforming standard SAEs (median AUROC of 0.80). This demonstrates its superior ability to capture functional motifs across diverse benchmarks.
4. MotifAE not only identifies motifs but also aligns with experimental data through gated feature selection, identifying features associated with specific properties such as folding stability. This alignment enhances performance in fitness prediction and enables the design of proteins with enhanced stability.
5. The study further demonstrates that MotifAE captures known functional motifs from the ELM database, with some features showing high specificity for certain motifs while others represent more general patterns. This versatility makes MotifAE a powerful tool for large-scale motif discovery.
6. MotifAE’s ability to capture homodimerization interfaces and align with three-dimensional functional sites highlights its potential for uncovering structural motifs. This capability is crucial for understanding protein-protein interactions and complex formation.
7. The authors developed MotifAE-G, a framework that integrates MotifAE with experimental data to identify features associated with specific functions. This approach significantly improves prediction performance on protein stability and provides a method for rational protein design.
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/CHAOHOU-97/MotifA…
#MotifAE #ProteinMotifs #SparseAutoencoder #ProteinLanguageModel #ESM2 #UnsupervisedLearning #ProteinEngineering #Bioinformatics
2
4
21
2,436
6 Nov 2025
Fine-tuned protein language model identifies antigen-specific B cell receptors from immune repertoires
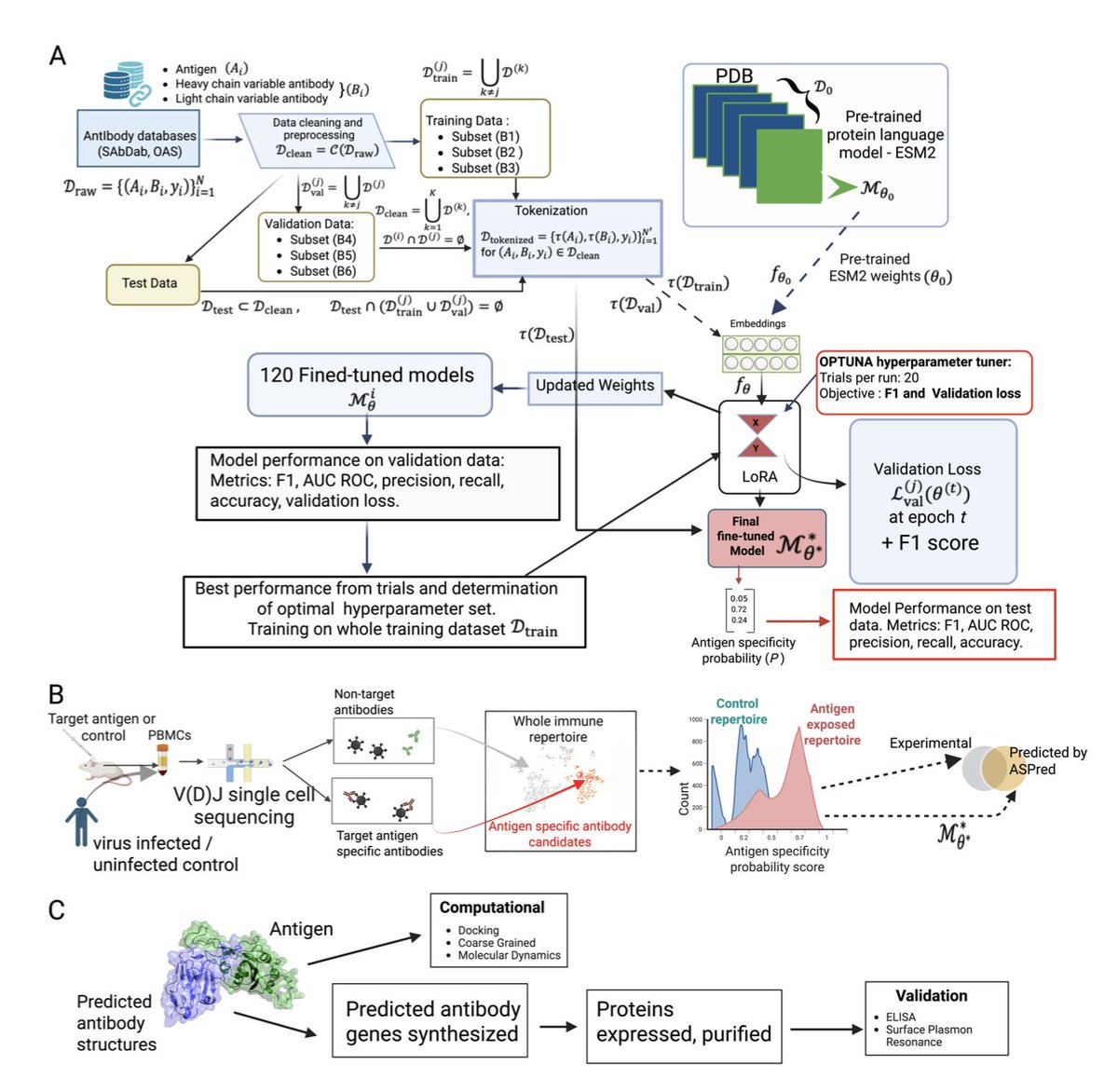
1. A new study introduces ASPred, a protein language model fine-tuned to predict antigen-specific antibodies directly from immune repertoire sequences. This innovation could revolutionize the scalability of antibody discovery and accelerate the development of countermeasures against emerging pathogens.
2. ASPred leverages fine-tuning of the ESM-2 protein language model on antibody heavy-chain sequences, achieving high accuracy in predicting antigen specificity for SARS-CoV-2, influenza, and HIV. The model’s ability to generalize across different antigens highlights its potential for broad application in immunology.
3. The study validates ASPred’s predictions through experimental binding assays and molecular dynamics simulations, demonstrating the model’s capacity to identify functional antibodies without prior selection. This approach significantly reduces the need for labor-intensive experimental methods.
4. ASPred’s predictions show significant overlap with experimental data from Barcode Enabled Antigen Mapping (BEAM), suggesting that the model can effectively identify antigen-specific B cell receptors from unselected immune repertoires. This scalability is a major step forward in computational immunology.
5. The model’s ability to identify diverse antibody sequences beyond simple clonal expansion indicates that it captures underlying biophysical principles of antigen recognition. This capability could lead to the discovery of novel antibodies with therapeutic potential.
6. ASPred’s framework integrates molecular simulations, transcriptomic analysis, and experimental validation, providing a comprehensive approach to understanding adaptive immunity. The study also highlights the potential for leveraging natural immune repertoires to develop safer and more effective biologics.
📜Paper: biorxiv.org/content/10.1101/…
#AntibodyDiscovery #ProteinLanguageModel #ComputationalImmunology #AIinBiology #AntigenSpecificity
2
14
1,356
4 Nov 2025
Structure-based Predictions of Conformational B Cell Epitopes by Protein Language Model and Deep Learning
1. A novel computational framework for predicting conformational B-cell epitopes has been introduced, leveraging protein language models (PLMs) and deep learning. This method represents a significant advancement in the field of epitope prediction, offering a more accurate and efficient alternative to traditional experimental and computational approaches.
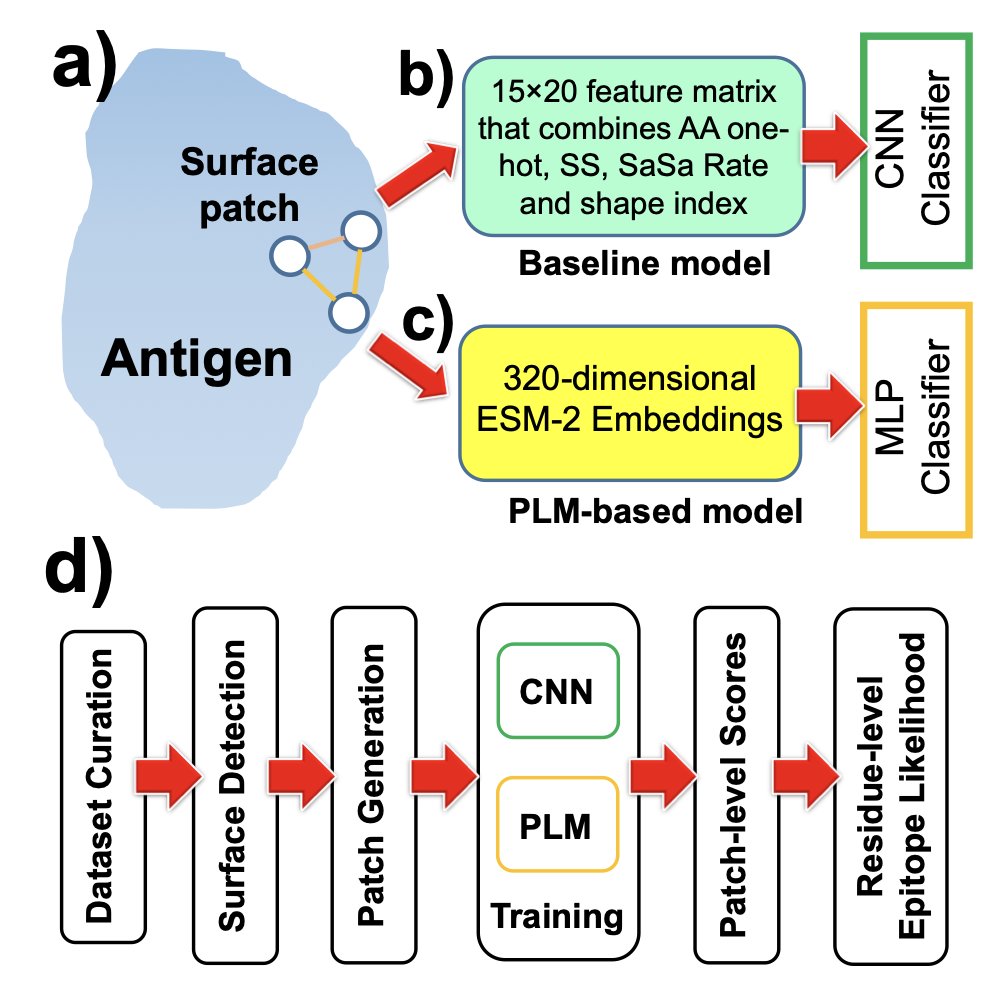
2. The core innovation lies in the use of a patch-centric approach, where each surface “patch” is defined as a triad of neighboring residues. This captures the smallest local unit that encodes both shape and chemistry, providing a detailed and nuanced representation of the antigen surface.
3. Two classifiers were evaluated: a PLM approach that averages ESM-2 embeddings over each triad and scores them with a small multilayer perceptron, and a convolutional baseline that uses a hand-crafted 15×20 feature matrix summarizing amino-acid identity, secondary structure, solvent accessibility, and shape index. The PLM model markedly outperformed the CNN at the patch level.
4. The PLM model achieved an F1 score of approximately 0.986 and an ROC–AUC of approximately 0.998, demonstrating superior performance in identifying epitopes. When aggregating patch scores to residues, the PLM model also surpassed the CNN in residue-wise performance, with an ROC–AUC of 0.689±0.072 compared to 0.548±0.018.
5. The method converts PLM representations into interpretable epitope likelihood maps, which can be used for antigen prioritization, antibody engineering, and vaccine design. This practical application makes the approach highly valuable for both research and clinical settings.
6. The study utilized a curated collection of 1,151 non-redundant antibody–antigen complexes from the AbDb database for training and validation. The PLM model achieved the best summary metrics on this benchmark, with an ROC–AUC of 0.67 and a PR–AUC of 0.56, outperforming widely used sequence and structure-based tools.
7. The model’s generalizability was tested on five external antibody–antigen complexes not used in development, and it performed well, with an ROC–AUC of 0.663 and accurate localization of binding regions. This indicates that the method can be effectively applied to new and unseen data.
📜Paper: biorxiv.org/content/10.1101/…
#EpitopePrediction #ProteinLanguageModel #DeepLearning #AntibodyAntigenInteraction #ComputationalBiology
1
4
13
1,767