
5 Jun 2025
AlphaFold 3 accurately models natural variants of Helicobacter pylori catalase KatA
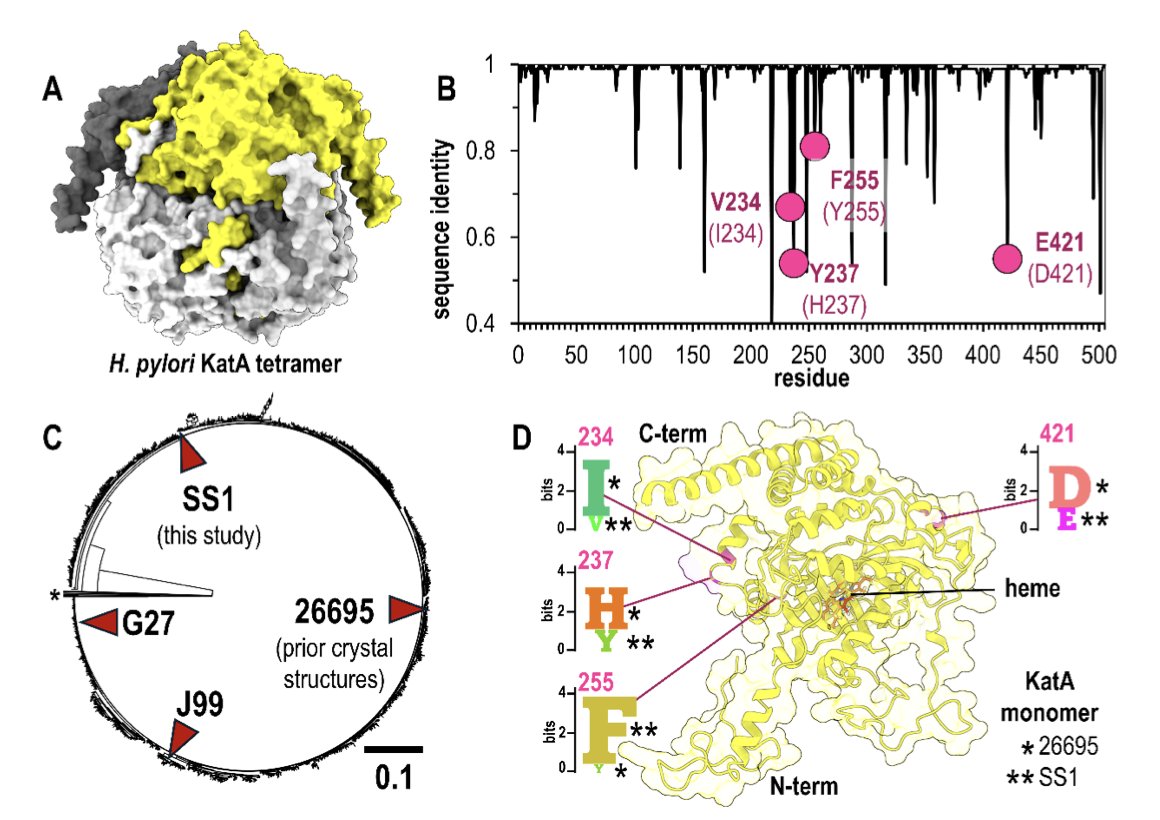
1.This study rigorously evaluates AlphaFold 3's ability to model natural protein variants using a high-resolution crystal structure of the H. pylori catalase KatA from strain SS1. These variants differ from the canonical 26695 strain at key residues: 234, 237, 255, and 421.
2.The most striking result: AlphaFold 3 accurately predicted both the global architecture and fine structural features of KatASS1, including conservative substitutions and solvent-exposed or interface residues—without access to the variant structure during training.
3.Variant residues modeled by AlphaFold 3 closely matched those resolved in the crystal structure (1.87 Å resolution). For Val234 and Phe255, the predictions reached experimental quality. For the more challenging Glu421 and Tyr237, performance varied by input.
4.Incorrect oligomeric state input (e.g., monomer instead of tetramer) reduced accuracy, particularly at interface-exposed residues like Glu421, which lost native hydrogen bonding seen in the crystal structure. Models with incorrect input can show misleadingly high pLDDT scores.
5.Minor input perturbations like single-residue substitutions or terminal Trp insertions had negligible impact on structure prediction, reinforcing the model's robustness—so long as the correct oligomeric state is used.
6.Among natural KatA variants from 1,931 H. pylori genomes, the four studied positions are common divergence points. Their accurate modeling suggests AlphaFold 3 may be broadly useful in evaluating natural variation in pathogen proteins with experimental backbones.
7.The authors highlight that AlphaFold 3’s accessibility is a double-edged sword—non-expert users may unintentionally degrade prediction quality by providing incorrect inputs. Even confident predictions (e.g., high pLDDT) can be structurally misleading.
8.This case study underscores AlphaFold 3's strong potential for modeling natural variants, especially when paired with known oligomeric context and conservative substitutions—but also highlights the need for caution and biological context in interpretation.
9.The experimental crystal structure of KatASS1, solved here for the first time, provides a new reference for benchmarking modeling of natural variants and has been deposited under PDB ID 9nh3.
📜Paper: biorxiv.org/content/10.1101/…
#AlphaFold3 #ProteinStructure #StructuralBiology #HelicobacterPylori #ComputationalBiology #ProteinVariants #PathogenEvolution

1
2
669
5 Jun 2025
AlphaFold 3 accurately models natural variants of Helicobacter pylori catalase KatA
1.This study rigorously evaluates AlphaFold 3's ability to model natural protein variants using a high-resolution crystal structure of the H. pylori catalase KatA from strain SS1. These variants differ from the canonical 26695 strain at key residues: 234, 237, 255, and 421.
2.The most striking result: AlphaFold 3 accurately predicted both the global architecture and fine structural features of KatASS1, including conservative substitutions and solvent-exposed or interface residues—without access to the variant structure during training.
3.Variant residues modeled by AlphaFold 3 closely matched those resolved in the crystal structure (1.87 Å resolution). For Val234 and Phe255, the predictions reached experimental quality. For the more challenging Glu421 and Tyr237, performance varied by input.
4.Incorrect oligomeric state input (e.g., monomer instead of tetramer) reduced accuracy, particularly at interface-exposed residues like Glu421, which lost native hydrogen bonding seen in the crystal structure. Models with incorrect input can show misleadingly high pLDDT scores.
5.Minor input perturbations like single-residue substitutions or terminal Trp insertions had negligible impact on structure prediction, reinforcing the model's robustness—so long as the correct oligomeric state is used.
6.Among natural KatA variants from 1,931 H. pylori genomes, the four studied positions are common divergence points. Their accurate modeling suggests AlphaFold 3 may be broadly useful in evaluating natural variation in pathogen proteins with experimental backbones.
7.The authors highlight that AlphaFold 3’s accessibility is a double-edged sword—non-expert users may unintentionally degrade prediction quality by providing incorrect inputs. Even confident predictions (e.g., high pLDDT) can be structurally misleading.
8.This case study underscores AlphaFold 3's strong potential for modeling natural variants, especially when paired with known oligomeric context and conservative substitutions—but also highlights the need for caution and biological context in interpretation.
9.The experimental crystal structure of KatASS1, solved here for the first time, provides a new reference for benchmarking modeling of natural variants and has been deposited under PDB ID 9nh3.
📜Paper: biorxiv.org/content/10.1101/…
#AlphaFold3 #ProteinStructure #StructuralBiology #HelicobacterPylori #ComputationalBiology #ProteinVariants #PathogenEvolution
9
21
1,110

1 Apr 2025
2/ Take a virus’s or bacteria's DNA or RNA. It’s a tiny string of letters. They are aligned against others to spot changes, like how SARS-CoV-2 evolved. That’s phylogenetics. Like mapping a family tree.
#PathogenEvolution
1
2
22

2 Dec 2024
Get to know our newly appointed Assistant Professor of Pathogen Evolution: Barbora Trubenová! Barbora is dedicated to understanding how bacteria, fungi, and parasitic worms develop resistance to drugs. 💊🧬👩🏻🔬
usys.ethz.ch/en/news-events/… @BTrubenova #PathogenEvolution
2
2
190

6 Oct 2024
skalo: using SKA split k-mers with coloured de Brujin graphs to genotype indels. #Indels #kmers #MycobacteriumData #PathogenEvolution @biorxivpreprint
biorxiv.org/content/10.1101/…
2
12
1,193

27 Aug 2023
[#phimaward] Meet #ElisabethFournier, expert in #Epidemiology #PathogenEvolution in #rice #agroecosystems #ICPP2023 @INRAE_DPT_SPE
26 Aug 2023

Fascinating team presentation by Frederic Suffert @wheatpath & Elisabeth Fournier @INRAE_France on epidemiological & evolutionary pathogen dynamics in cultivar mixtures in #wheat & #rice spanning short to long timescales #ICPP2023



2
6
741

3 Jul 2023
(2/2)
We were also encouraged to initiate an experiment where we test the concepts we learn!Excited to see the outcomes! (Pic: Me sitting in a hood after 3 years!)
#pathogenevolution #microbiology

4
136

24 Jun 2023
Had a fascinating session with Prof. Rahul Roy discussing the research carried out by his group. Combining experimental and modeling approaches, he sheds light on pathogen evolution over decades. Mind-blowing insights! #BiologicalReactions #PathogenEvolution #ICES23

13
356

#OneHealthMedia
15min @OneHealthTrust podcast episode on Lemurs in Madagascar 'When helping animals means helping people, too'
Great work @PlanetMada !
#Conservation #Primates #Cryptosporidiosis #SIV #PathogenEvolution #BioAnthropology #sifaka
8 Feb 2023

Dr. Travis Steffens @GuelphSOAN stars in the latest One Health Trust, One World One Health podcast with @maggiemfox!: tinyurl.com/4jzdh7k8
She spoke with Travis about his work to save Madagascar's lemurs and improve the lives of people who live with and near them.
#OneHealth

ALT A blue and green graphic promoting a One World One Health podcast by the One Health Trust entitled "Leaping Lemurs; When helping animals means helping people too" with special guest Dr. Travis Steffens. The graphic also features an illustration of a lemur jumping through the air.
5
173

10 Mar 2022
Now available: academic.oup.com/past/advanc…. #histmed #plague #2ndPlaguePandemic #aDNA #PathogenEvolution A thread summarizing will follow.
1
3

4 Feb 2022
Happy to announce the publication of our latest paper revealing, “A role for ColV plasmids in the evolution of pathogenic E. coli ST58” in @NatureComms. Check out this thread for the TL;DR @maxcummins1 @steve_djo #pathogenevolution #microbialgenomics nature.com/articles/s41467-0…
4
10
27

17 May 2021
Review article: The population genomics of within-host Mycobacterium tuberculosis go.nature.com/3kooct5
#Mycobacterium #populationGenomics #withinhost #resistanceEvolution #pathogenEvolution
1
4
12 Apr 2021
Review article: The population genomics of within-host Mycobacterium tuberculosis go.nature.com/3kooct5
#Mycobacterium #populationGenomics #withinhost #resistanceEvolution #pathogenEvolution
1
2

1 Apr 2021
POINT OF NO RETURN Ex-CDC Boss: Covid Came From Lab!; Vaccine Passports & the Last Step to Tyranny?; Science Shows Vaccines Can Make Pathogens Deadlier; “Covid Crazy” Judge Targets FL Mom #EndofAmerica #PathogenEvolution #CovidOrigin #Covid19 pscp.tv/w/czSwKzFETEVCeU1Xb0…
44
277
498
9 Mar 2021
Review article: The population genomics of within-host Mycobacterium tuberculosis go.nature.com/3kooct5
#Mycobacterium #populationGenomics #withinhost #resistanceEvolution #pathogenEvolution
1
2
6 Feb 2021
Review article: The population genomics of within-host Mycobacterium tuberculosis go.nature.com/3kooct5
#Mycobacterium #populationGenomics #withinhost #resistanceEvolution #pathogenEvolution
1

27 Jan 2021
Better genetic surveillance will help control the pandemic. So, why are many countries doing a poor job at tracking COVID19 mutations?
#WHO #VirusMutations #COVID19 #UKVariant #PathogenEvolution
sciencealert.com/many-countr…
2
6

1 Dec 2020
a great work: nextstrain.org/ for
"Real-time tracking of #pathogenEvolution
#Nextstrain is an #opensource project to harness the scientific and public health potential of pathogen genome data."
3

28 May 2019
Over time, environmental forces induce ID gene change. Host defenses R restricted by unhealthy people. Irresponsibly administered vaccine health policy w/intent 2 control dangerous diseases can perversely predispose compromised individuals. #immuneevasion #pathogenevolution

5 Feb 2019
Awesome v2. Thanks to #TWIV for reading out my letter on ep 533 bit.ly/2D82ea7, in reply to last week's discussion about morbilliviruses and our paper. #pathogenevolution #rinderpest. Really cool to hear even more discussion on this topic. #lovevirology

New episode of the #science #podcast This Week in Virology #TWiV 533: Recurring threads bit.ly/2D82ea7 Chinese plasma virome revealed by non-invasive prenatal testing, and a new filovirus genome from bats in China with @BioProfBarker @RichCondit @VirusProfSings
6