
15 Oct 2025
Protein as a Second Language for LLMs
1. A novel study has introduced a novel framework that enables large language models (LLMs) to interpret protein sequences as a symbolic language, significantly enhancing their ability to understand and predict protein functions without task-specific fine-tuning.
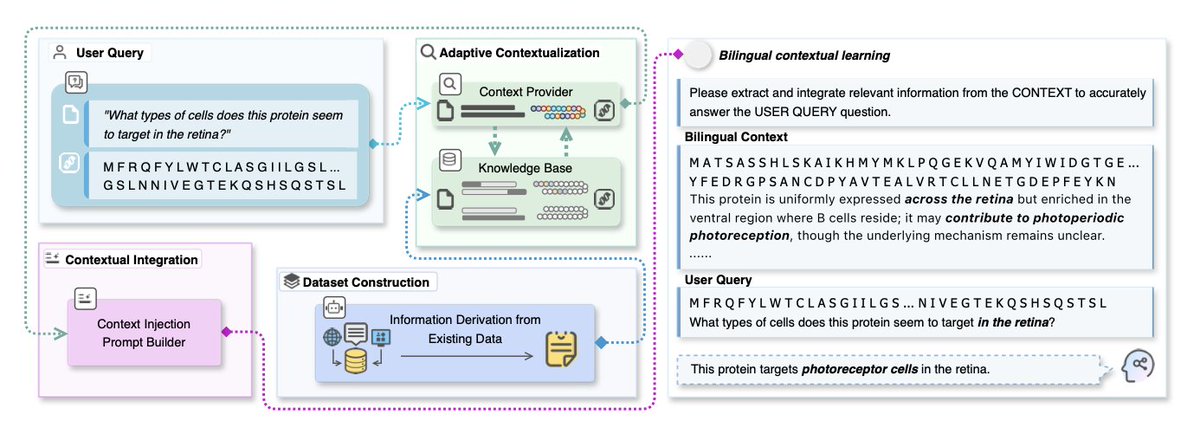
2. The core innovation lies in treating protein sequences as a "second language" that LLMs can learn through contextual examples. This approach constructs sequence–question–answer triples that reveal functional cues, allowing LLMs to acquire protein semantics in a zero-shot setting.
3. The researchers curated a bilingual dataset of 79,926 protein–QA instances, covering attribute prediction, descriptive understanding, and extended reasoning. This dataset supports effective learning and benchmarking for protein language models.
4. Empirical results show that this method delivers consistent gains across diverse open-source LLMs and GPT-4o, achieving up to 17.2% ROUGE-L improvement and even surpassing fine-tuned protein-specific language models.
5. The adaptive context construction mechanism selectively builds bilingual learning contexts for each query, exposing LLMs to new words in context so that meaning and usage can be inferred, similar to how humans acquire a second language.
6. The study demonstrates that general-purpose LLMs, when guided with protein-as-language cues, can outperform domain-specialized models, offering a scalable pathway for protein understanding.
7. The framework has been evaluated on multiple protein-language datasets, showing significant improvements over zero-shot baselines and highlighting the effectiveness of context-driven learning in bridging protein sequences with functional descriptions.
📜Paper: arxiv.org/abs/2510.11188v1
#ProteinLanguage #LLMs #Bioinformatics #AIinBiology #ProteinFunctionPrediction
2
17
1,613
30 Sep 2025
Predicting peptide aggregation with protein language model embeddings
1. A new deep-learning model named PALM (Predicting Aggregation with Language Model embeddings) has been developed to predict peptide aggregation, which is associated with multiple diseases and hinders the development of therapeutics. This model leverages transfer learning from pretrained protein language model (pLM) embeddings, showing strong performance on held-out experimental datasets.
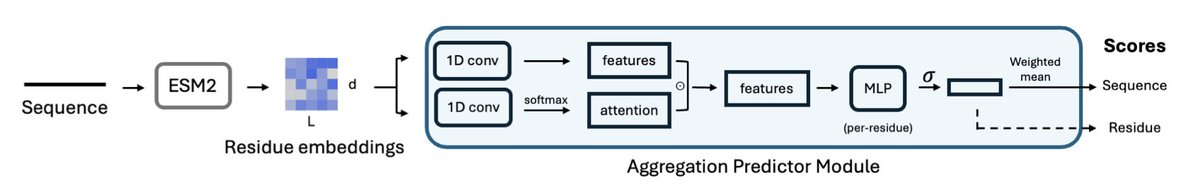
2. PALM is trained on WaltzDB-2.0 and uses embeddings from the pretrained ESM2 model. It employs an adapted Light Attention architecture called the Aggregation Predictor Module (APM) to extract local sequence patterns and predict aggregation at single-residue resolution. The key innovation is that the APM infers residue-specific contributions from experimental data with sequence-level annotations.
3. The study finds that PALM fails to identify single mutations that increase the rate of aggregation of amyloid beta peptide. However, training the PALM architecture on a larger dataset, CANYA NNK1-3, substantially improves performance in this task. This highlights that challenging tasks, such as predicting the effect of single mutations, require more experimental data.
4. The model's performance is evaluated on multiple datasets. It shows higher similarity to Serrano158 and AmyPro22 when WaltzDB sequences are padded with additional residues. The smallest ESM2 model provides the best performance, and the APM enables residue-level prediction of aggregation-promoting regions.
5. PALM demonstrates strong performance compared to existing models on both sequence-level and residue-level aggregation prediction tasks. It also shows potential for identifying aggregation-prone regions within protein sequences without being explicitly trained on this task. The study concludes that PALM can be improved with more data and has implications for therapeutic peptide development.
📜Paper: biorxiv.org/content/10.1101/…
#peptideaggregation #proteinlanguage #deeplearning #bioinformatics #therapeuticdevelopment
1
5
988
25 Apr 2025
Inferring context-specific site variation with evotuned protein language models
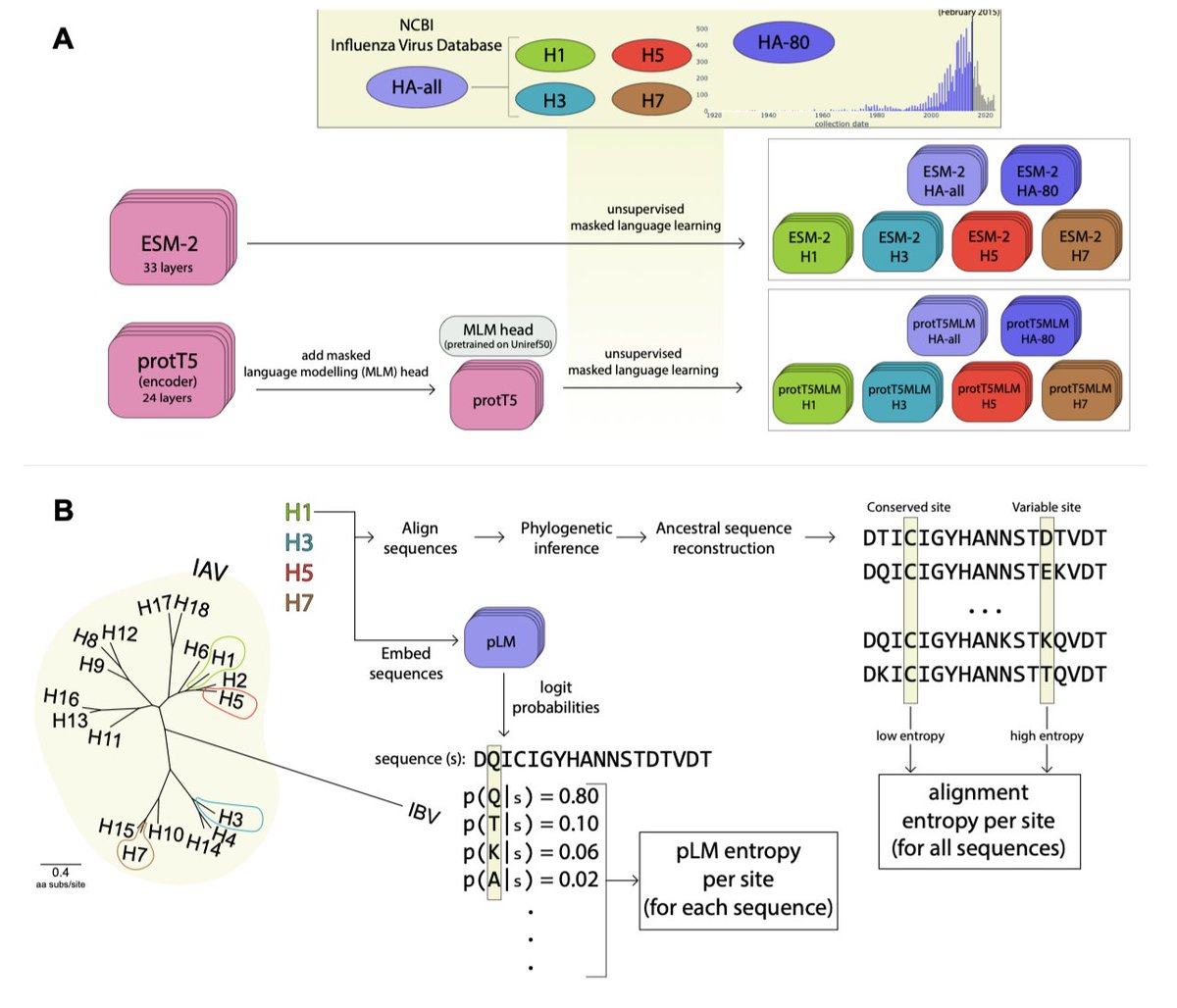
1/ In this study, the authors introduce a novel metric, 'pLM entropy', to assess protein site conservation and variability, based on protein language models (pLMs) rather than traditional sequence alignments.
2/ The pLM entropy approach offers a unique advantage over multiple sequence alignments (MSAs) by enabling site-specific inferences from single sequences, making it particularly useful for studying proteins with limited or no aligned sequence data.
3/ The study demonstrates how fine-tuning (evotuning) pre-existing models, such as ESM-2 and protT5, on virus-specific protein data (like Influenza A Hemagglutinin) enhances their ability to predict the variability and conservation of specific sites across related proteins.
4/ A key finding is that pLM entropy effectively captures which sites are likely to change in a given sequence context, which is crucial for understanding protein evolution and potentially predicting mutation hotspots.
5/ By comparing the predictive power of evotuned models versus alignment-based metrics, the authors show that pLM entropy improves predictions of evolutionary changes at specific sites, surpassing traditional alignment entropy in predictive tasks.
6/ The study also explores how model performance varies with dataset characteristics, noting that even with smaller or less genetically related datasets, evotuned pLMs maintain robust performance, highlighting the method's versatility and adaptability.
7/ This research opens new possibilities for applying pLMs in viral evolution studies, offering a potentially powerful tool for tracking pathogen evolution and anticipating future mutations, especially in preparation for pandemics.
📜Paper: biorxiv.org/content/10.1101/…
#proteinlanguage #machinelearning #bioinformatics #evolutionarybiology #pLM #Influenza #virus
3
8
841
25 Apr 2025
Inferring context-specific site variation with evotuned protein language models
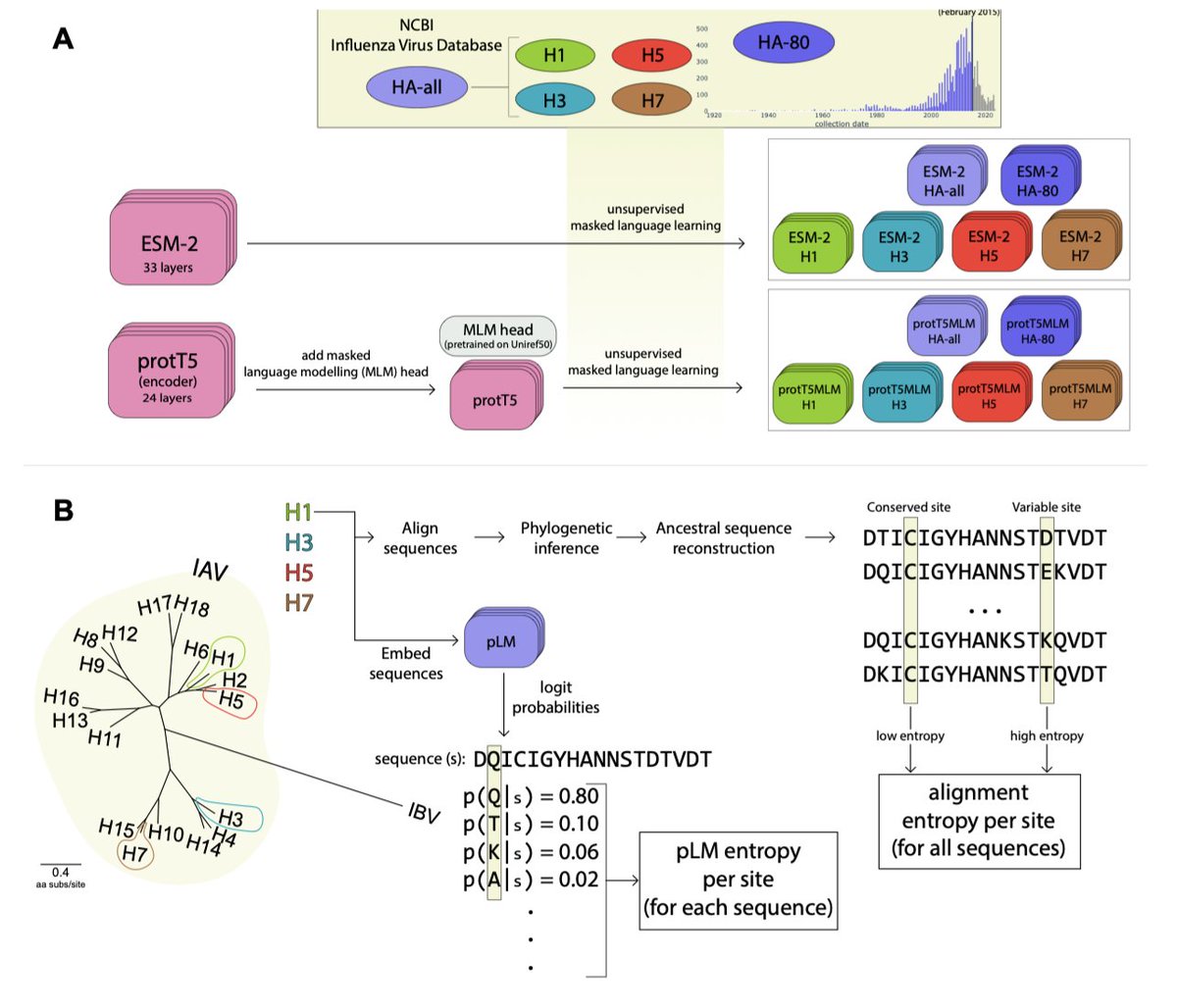
1/ In this study, the authors introduce a novel metric, 'pLM entropy', to assess protein site conservation and variability, based on protein language models (pLMs) rather than traditional sequence alignments.
2/ The pLM entropy approach offers a unique advantage over multiple sequence alignments (MSAs) by enabling site-specific inferences from single sequences, making it particularly useful for studying proteins with limited or no aligned sequence data.
3/ The study demonstrates how fine-tuning (evotuning) pre-existing models, such as ESM-2 and protT5, on virus-specific protein data (like Influenza A Hemagglutinin) enhances their ability to predict the variability and conservation of specific sites across related proteins.
4/ A key finding is that pLM entropy effectively captures which sites are likely to change in a given sequence context, which is crucial for understanding protein evolution and potentially predicting mutation hotspots.
5/ By comparing the predictive power of evotuned models versus alignment-based metrics, the authors show that pLM entropy improves predictions of evolutionary changes at specific sites, surpassing traditional alignment entropy in predictive tasks.
6/ The study also explores how model performance varies with dataset characteristics, noting that even with smaller or less genetically related datasets, evotuned pLMs maintain robust performance, highlighting the method's versatility and adaptability.
7/ This research opens new possibilities for applying pLMs in viral evolution studies, offering a potentially powerful tool for tracking pathogen evolution and anticipating future mutations, especially in preparation for pandemics.
📜Paper: biorxiv.org/content/10.1101/…
#proteinlanguage #machinelearning #bioinformatics #evolutionarybiology #pLM #Influenza #virus
6
610
17 Apr 2025
A Benchmarking Platform for Assessing Protein Language Models on Function-related Prediction Tasks
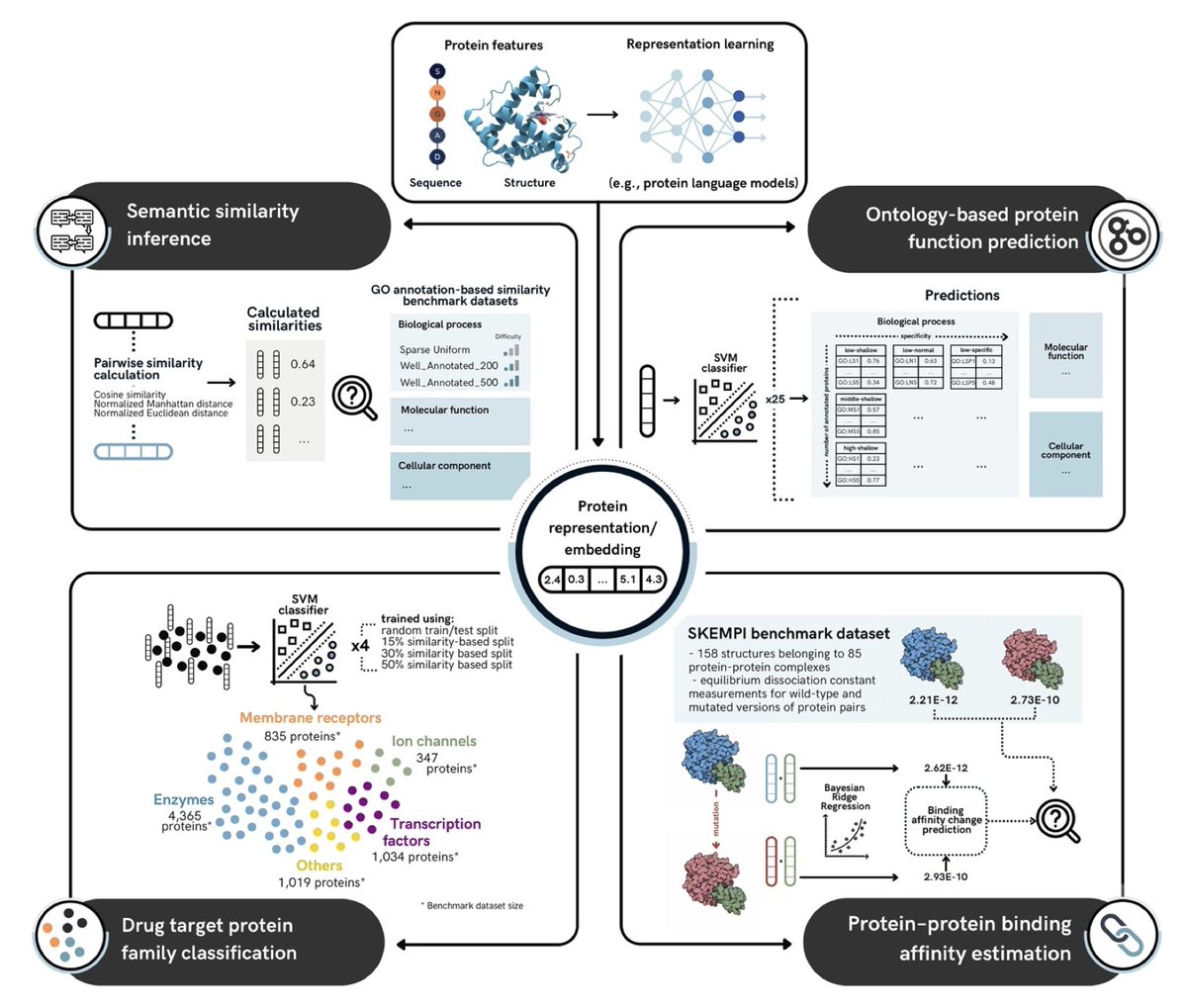
1. The study introduces the Protein Representation Benchmark (PROBE), a comprehensive framework for evaluating protein language models (PLMs) on four key function-related prediction tasks: semantic similarity inference, ontology-based protein function prediction, drug target family classification, and protein-protein binding affinity estimation.
2. PROBE is designed to assess how well protein representations, from classical methods to state-of-the-art PLMs, capture and predict functional characteristics of proteins, offering a comparative platform for both existing and newly developed models.
3. The framework includes a user-friendly interface and can process protein embeddings from diverse sources, making it accessible to a wide range of researchers working on protein function prediction, drug discovery, and protein interaction studies.
4. The study highlights the performance of various PLMs, including ESM2, ESM3, ProstT5, and SaProt, showing how multimodal models that incorporate both sequence and structural data outperform traditional methods in several tasks, particularly in semantic similarity and function prediction.
5. In the ontology-based protein function prediction task, the multimodal models (ESM3, ProstT5) performed particularly well, achieving high accuracy in predicting Gene Ontology (GO) terms across molecular function, biological process, and cellular component categories.
6. For drug target classification, ProtT5-XL led the benchmark in predicting the correct family of drug target proteins, highlighting the model's ability to capture evolutionary and functional patterns critical for therapeutic targeting.
7. In protein-protein binding affinity prediction, ProtALBERT showed superior performance, outclassing traditional models and indicating that transformer-based models with attention mechanisms are particularly effective for capturing amino acid interactions.
8. The benchmarking results provide critical insights into the trade-offs between different protein representation methods and how they can be optimized for various functional prediction tasks, offering a valuable tool for PLM developers.
9. PROBE is not only a tool for evaluating current models but also serves as a resource for guiding future developments in protein function prediction, facilitating the integration of multimodal data into PLM training.
10. This work emphasizes the importance of rigorous, task-specific benchmarking in advancing protein representation models and enhancing the prediction of functional protein characteristics, which is vital for drug discovery and protein engineering.
💻Code: github.com/kansil/PROBE
📜Paper: biorxiv.org/content/10.1101/…
#proteinfunction #bioinformatics #proteinlanguage #deeplearning #PLMs #drugdiscovery #proteinrepresentation #AI4Science #machinelearning #multimodalmodels
3
11
47
3,971
17 Apr 2025
A Benchmarking Platform for Assessing Protein Language Models on Function-related Prediction Tasks
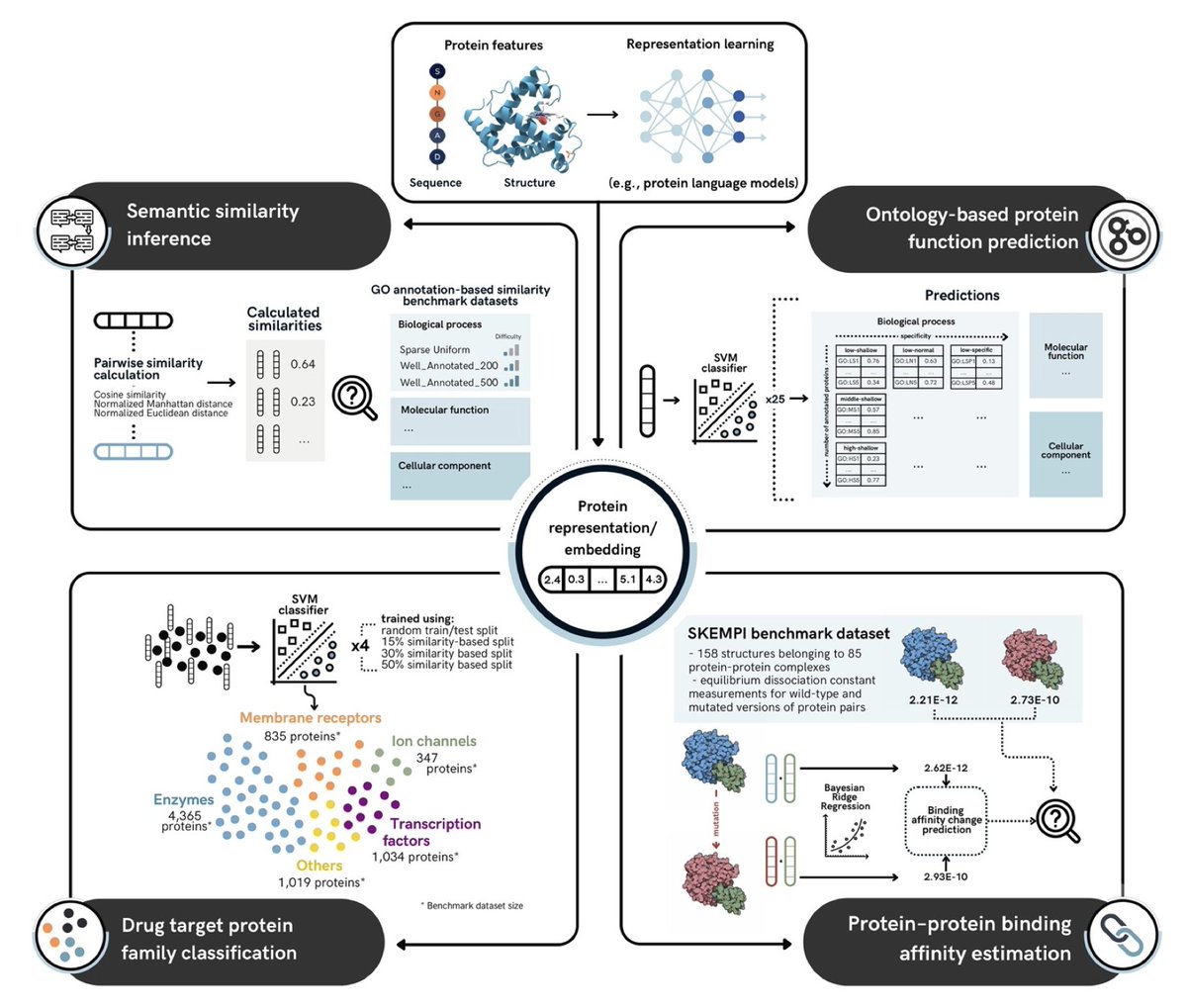
1. The study introduces the Protein Representation Benchmark (PROBE), a comprehensive framework for evaluating protein language models (PLMs) on four key function-related prediction tasks: semantic similarity inference, ontology-based protein function prediction, drug target family classification, and protein-protein binding affinity estimation.
2. PROBE is designed to assess how well protein representations, from classical methods to state-of-the-art PLMs, capture and predict functional characteristics of proteins, offering a comparative platform for both existing and newly developed models.
3. The framework includes a user-friendly interface and can process protein embeddings from diverse sources, making it accessible to a wide range of researchers working on protein function prediction, drug discovery, and protein interaction studies.
4. The study highlights the performance of various PLMs, including ESM2, ESM3, ProstT5, and SaProt, showing how multimodal models that incorporate both sequence and structural data outperform traditional methods in several tasks, particularly in semantic similarity and function prediction.
5. In the ontology-based protein function prediction task, the multimodal models (ESM3, ProstT5) performed particularly well, achieving high accuracy in predicting Gene Ontology (GO) terms across molecular function, biological process, and cellular component categories.
6. For drug target classification, ProtT5-XL led the benchmark in predicting the correct family of drug target proteins, highlighting the model's ability to capture evolutionary and functional patterns critical for therapeutic targeting.
7. In protein-protein binding affinity prediction, ProtALBERT showed superior performance, outclassing traditional models and indicating that transformer-based models with attention mechanisms are particularly effective for capturing amino acid interactions.
8. The benchmarking results provide critical insights into the trade-offs between different protein representation methods and how they can be optimized for various functional prediction tasks, offering a valuable tool for PLM developers.
9. PROBE is not only a tool for evaluating current models but also serves as a resource for guiding future developments in protein function prediction, facilitating the integration of multimodal data into PLM training.
10. This work emphasizes the importance of rigorous, task-specific benchmarking in advancing protein representation models and enhancing the prediction of functional protein characteristics, which is vital for drug discovery and protein engineering.
💻Code: github.com/kansil/PROBE
📜Paper: biorxiv.org/content/10.1101/…
#proteinfunction #bioinformatics #proteinlanguage #deeplearning #PLMs #drugdiscovery #proteinrepresentation #AI4Science #machinelearning #multimodalmodels
6
772
17 Apr 2025
Learning Biophysical Dynamics with Protein Language Models
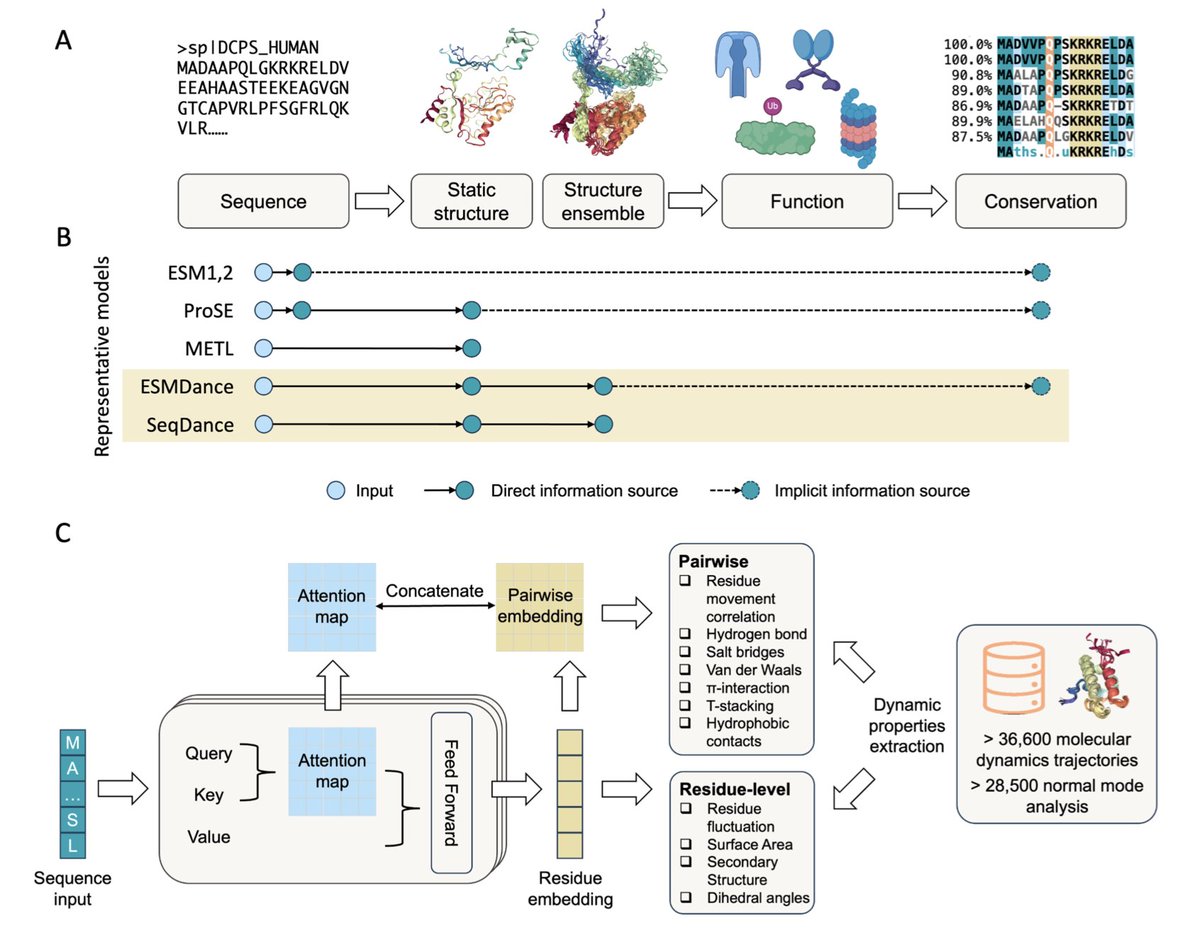
1. This work introduces SeqDance and ESMDance, two transformer-based protein language models trained on dynamic properties derived from molecular dynamics and normal mode analysis, offering a novel approach to capture protein dynamics directly from sequence.
2. Unlike conventional protein language models trained solely on evolutionary sequences, SeqDance learns from biophysical data without any structural or homolog information, enabling generalization to proteins with few or no known homologs, such as viral or de novo designed proteins.
3. ESMDance builds upon ESM2 and integrates both evolutionary and dynamic signals. It outperforms ESM2 and SeqDance in zero-shot prediction of mutation effects, achieving a median Spearman correlation of 0.46 on 412 proteins—matching or exceeding even ESM2-650M and ESM2-15B.
4. SeqDance captures both local residue interactions and global conformational properties like radius of gyration and asphericity. It consistently outperforms ProSE, METL, and ESM2 in predicting shape and flexibility of disordered and ordered proteins.
5. When predicting mutation-induced stability changes (ΔΔG), SeqDance shows clear correlations between predicted dynamic property shifts and experimental data, achieving zero-shot mutation effect predictions even for proteins outside its training set.
6. ESMDance excels particularly in designed and viral proteins where evolutionary-based models fail. On a set of 135 designed proteins with no homologs, it surpasses all ESM2 models and SeqDance, proving the strength of dynamics-aware modeling.
7. Attention map analysis reveals that SeqDance learns interpretable biophysical features like residue co-movement and interaction patterns, with high generalization to unseen simulation conditions and diverse protein classes including IDRs.
8. The authors curated dynamic properties for over 65,000 proteins, including all-atom and coarse-grained MD simulations and normal mode analysis data, spanning ordered domains, IDRs, membrane proteins, and complexes.
9. Training required only 1/1000th the number of sequences used for large PLMs like ESM2, demonstrating that rich physical signals can compensate for massive evolutionary datasets and lead to more mechanistic understanding.
10. By bridging sequence, dynamics, and function, SeqDance and ESMDance offer a powerful framework for rational protein design and mutation effect prediction, especially in contexts where evolutionary signals are sparse or misleading.
💻Code: github.com/ShenLab/SeqDance
📜Paper: biorxiv.org/content/10.1101/…
#proteinlanguage #bioinformatics #proteinengineering #moleculardynamics #mutationprediction #deeplearning #IDRs #computationalbiology #AI4Science #SeqDance #ESMDance
17
85
6,941
17 Apr 2025
Learning Biophysical Dynamics with Protein Language Models
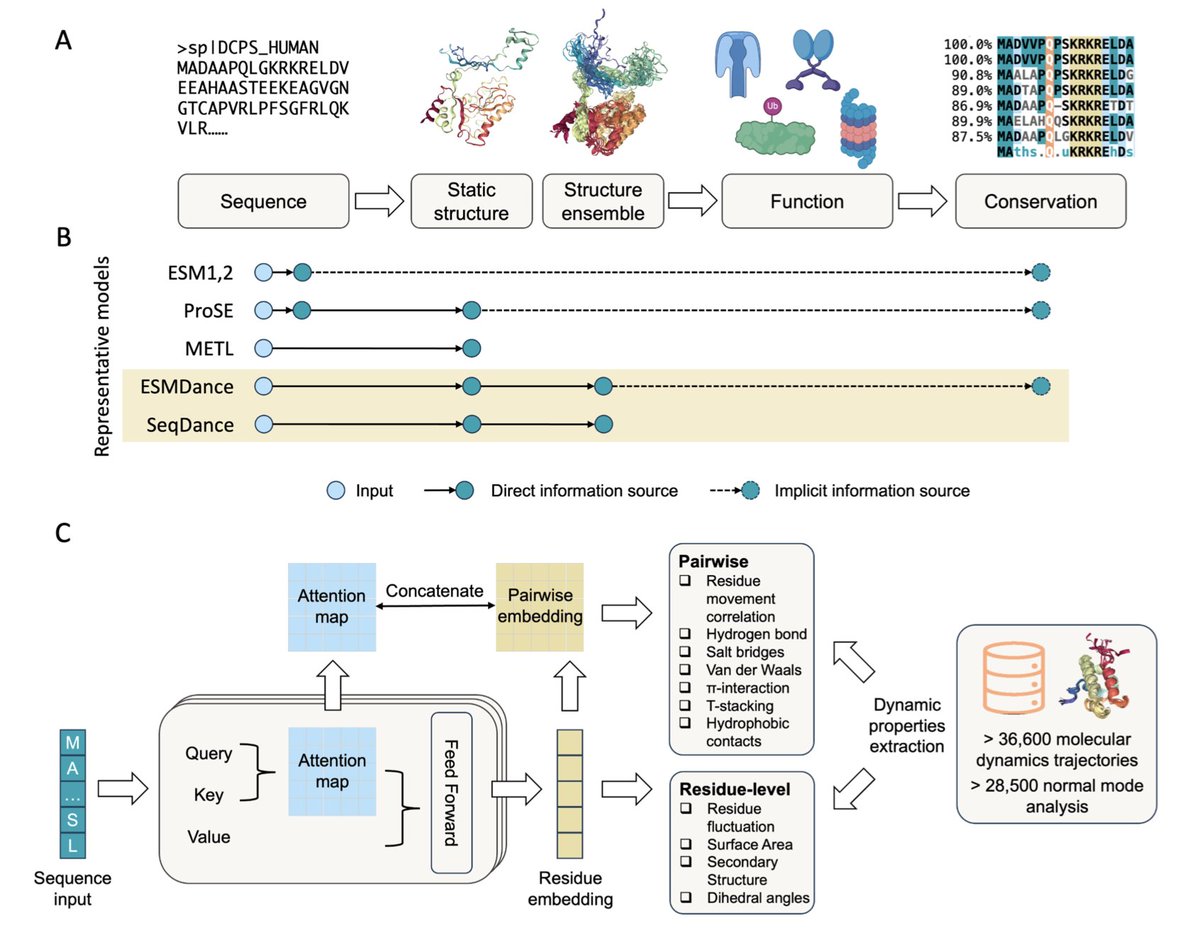
1. This work introduces SeqDance and ESMDance, two transformer-based protein language models trained on dynamic properties derived from molecular dynamics and normal mode analysis, offering a novel approach to capture protein dynamics directly from sequence.
2. Unlike conventional protein language models trained solely on evolutionary sequences, SeqDance learns from biophysical data without any structural or homolog information, enabling generalization to proteins with few or no known homologs, such as viral or de novo designed proteins.
3. ESMDance builds upon ESM2 and integrates both evolutionary and dynamic signals. It outperforms ESM2 and SeqDance in zero-shot prediction of mutation effects, achieving a median Spearman correlation of 0.46 on 412 proteins—matching or exceeding even ESM2-650M and ESM2-15B.
4. SeqDance captures both local residue interactions and global conformational properties like radius of gyration and asphericity. It consistently outperforms ProSE, METL, and ESM2 in predicting shape and flexibility of disordered and ordered proteins.
5. When predicting mutation-induced stability changes (ΔΔG), SeqDance shows clear correlations between predicted dynamic property shifts and experimental data, achieving zero-shot mutation effect predictions even for proteins outside its training set.
6. ESMDance excels particularly in designed and viral proteins where evolutionary-based models fail. On a set of 135 designed proteins with no homologs, it surpasses all ESM2 models and SeqDance, proving the strength of dynamics-aware modeling.
7. Attention map analysis reveals that SeqDance learns interpretable biophysical features like residue co-movement and interaction patterns, with high generalization to unseen simulation conditions and diverse protein classes including IDRs.
8. The authors curated dynamic properties for over 65,000 proteins, including all-atom and coarse-grained MD simulations and normal mode analysis data, spanning ordered domains, IDRs, membrane proteins, and complexes.
9. Training required only 1/1000th the number of sequences used for large PLMs like ESM2, demonstrating that rich physical signals can compensate for massive evolutionary datasets and lead to more mechanistic understanding.
10. By bridging sequence, dynamics, and function, SeqDance and ESMDance offer a powerful framework for rational protein design and mutation effect prediction, especially in contexts where evolutionary signals are sparse or misleading.
💻Code: github.com/ShenLab/SeqDance
📜Paper: biorxiv.org/content/10.1101/…
#proteinlanguage #bioinformatics #proteinengineering #moleculardynamics #mutationprediction #deeplearning #IDRs #computationalbiology #AI4Science #SeqDance #ESMDance
1
15
66
3,326
16 Apr 2025
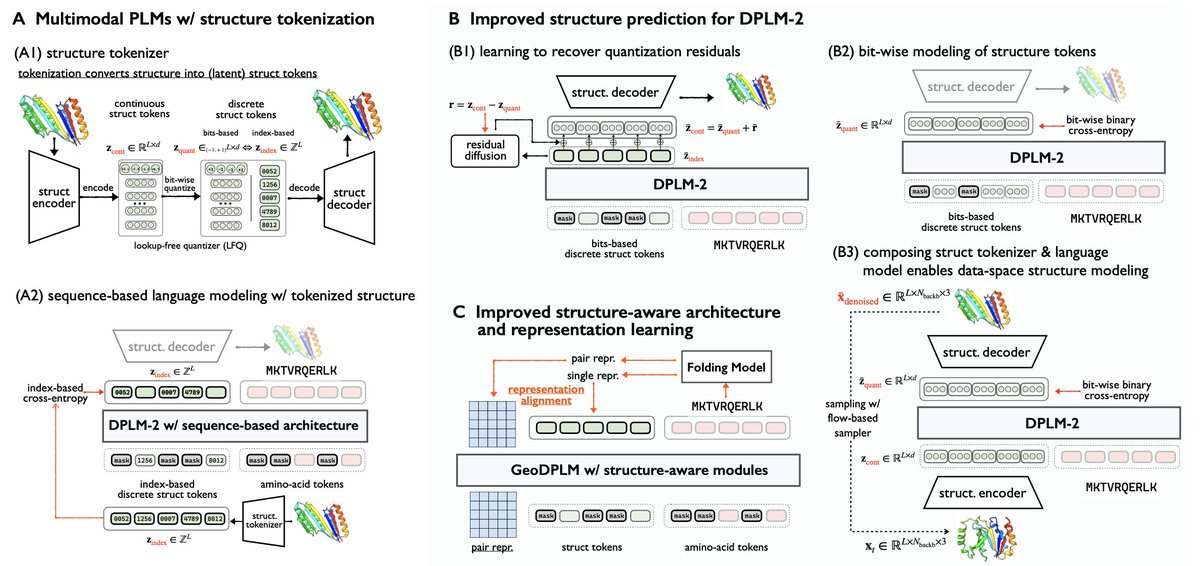
Elucidating the Design Space of Multimodal Protein Language Models
1. This paper presents a systematic exploration of the design space for multimodal protein language models (PLMs), identifying key limitations in token-based structural modeling and proposing architectural and training innovations to overcome them.
2. Tokenizing 3D protein structures into discrete tokens, a common practice in multimodal PLMs like DPLM-2 and ESM3, causes fidelity loss in structural detail. The authors show that bit-wise token representations and residual modeling can recover this loss and improve folding accuracy.
3. Bit-wise supervision significantly outperforms index-based structural tokens, increasing structure prediction accuracy while reducing deviation from ground truth. On PDB test sets, it reduces RMSD from 5.52 to 3.22 and increases TMscore to over 0.90.
4. The authors introduce RESDIFF, a lightweight residual diffusion module to recover fine-grained geometric details lost during tokenization. When combined with bit-based modeling, it further improves folding accuracy with minimal computational overhead.
5. A hybrid generative model is proposed, combining structure encoder, PLM, and decoder as a structure denoising pipeline. This enables direct data-space structure generation using flow matching, improving atomic-level predictions without sacrificing scalability.
6. To incorporate geometric inductive biases, the authors introduce GeoDPLM, which integrates structure-aware attention and pairwise representations, akin to AlphaFold’s PairFormer. These modules refine folding and improve generation diversity.
7. REPA (Representation Alignment to Folding Models) transfers structural semantics from pretrained folding models (e.g., ESMFold) to the PLM. This soft alignment boosts structural diversity and generative performance across architectures.
8. Combining bit-based modeling, geometric modules, and REPA achieves state-of-the-art folding accuracy with only 650M parameters, matching or surpassing 3B-size baselines like ESMFold and MultiFlow, while improving training speed and sample diversity.
9. The study also explores the impact of multimeric protein data. Incorporating multimer chains and position offsets into modeling improves performance for both multimer and monomer structures, indicating shared structural principles.
10. The final recommended setting—GeoDPLM with bit-based modeling—offers the best trade-off between accuracy, diversity, training efficiency, and unconditional generation, laying the foundation for future scalable and general-purpose protein models.
💻Code: bytedance.github.io/dplm/dpl…
📜Paper: arxiv.org/abs/2504.11454
#proteindesign #bioinformatics #proteinlanguage #multimodalmodels #deeplearning #proteinstructure #generativemodels #AlphaFold #AI4Science #diffusionmodels
3
8
46
6,671
16 Apr 2025
Elucidating the Design Space of Multimodal Protein Language Models
1. This paper presents a systematic exploration of the design space for multimodal protein language models (PLMs), identifying key limitations in token-based structural modeling and proposing architectural and training innovations to overcome them.
2. Tokenizing 3D protein structures into discrete tokens, a common practice in multimodal PLMs like DPLM-2 and ESM3, causes fidelity loss in structural detail. The authors show that bit-wise token representations and residual modeling can recover this loss and improve folding accuracy.
3. Bit-wise supervision significantly outperforms index-based structural tokens, increasing structure prediction accuracy while reducing deviation from ground truth. On PDB test sets, it reduces RMSD from 5.52 to 3.22 and increases TMscore to over 0.90.
4. The authors introduce RESDIFF, a lightweight residual diffusion module to recover fine-grained geometric details lost during tokenization. When combined with bit-based modeling, it further improves folding accuracy with minimal computational overhead.
5. A hybrid generative model is proposed, combining structure encoder, PLM, and decoder as a structure denoising pipeline. This enables direct data-space structure generation using flow matching, improving atomic-level predictions without sacrificing scalability.
6. To incorporate geometric inductive biases, the authors introduce GeoDPLM, which integrates structure-aware attention and pairwise representations, akin to AlphaFold’s PairFormer. These modules refine folding and improve generation diversity.
7. REPA (Representation Alignment to Folding Models) transfers structural semantics from pretrained folding models (e.g., ESMFold) to the PLM. This soft alignment boosts structural diversity and generative performance across architectures.
8. Combining bit-based modeling, geometric modules, and REPA achieves state-of-the-art folding accuracy with only 650M parameters, matching or surpassing 3B-size baselines like ESMFold and MultiFlow, while improving training speed and sample diversity.
9. The study also explores the impact of multimeric protein data. Incorporating multimer chains and position offsets into modeling improves performance for both multimer and monomer structures, indicating shared structural principles.
10. The final recommended setting—GeoDPLM with bit-based modeling—offers the best trade-off between accuracy, diversity, training efficiency, and unconditional generation, laying the foundation for future scalable and general-purpose protein models.
💻Code: bytedance.github.io/dplm/dpl…
📜Paper: arxiv.org/abs/2504.11454
#proteindesign #bioinformatics #proteinlanguage #multimodalmodels #deeplearning #proteinstructure #generativemodels #AlphaFold #AI4Science #diffusionmodels

2
5
22
2,908

21 Jan 2025
#InnovativeTsinghua researchers proposed CLAPE-SMB, which uses a #proteinlanguage model and contrastive learning to predict small #molecule binding sites without #crystal structures, advancing drug design and unraveling protein-small molecule interactions. bit.ly/4hWaqNj

3
1,119

9 Oct 2023
Sequence-based Protein-Protein Interaction Prediction Using Multi-kernel Deep Convolutional Neural Networks with ProteinLanguage Model biorxiv.org/cgi/content/shor… #bioRxiv
1
2,322

13 Jun 2023
#ProteinLanguage model to predict protein-membrane interfaces

13 Jun 2023

@ALLODD_ITN Leader @zoecournia presenting on Exploiting #Allostery for #CADD of #Oncogenes at #MedChemFrontiers2023 !

2
5
891