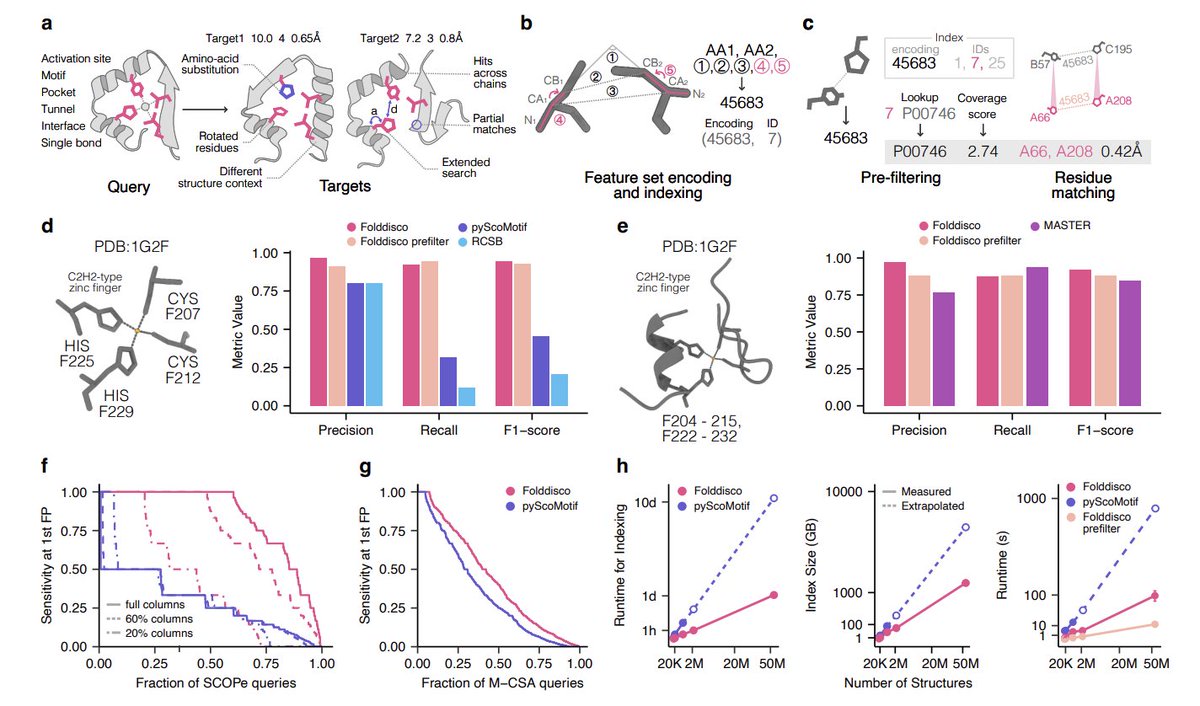
Structural motif search across the protein-universe with Folddisco @NatureBiotech
1 Folddisco is a structural-motif search engine designed for the “protein structure explosion” era: it can query 53 million clustered AlphaFoldDB structures (AFDB50) in seconds, enabling large-scale discovery of short 3D functional patterns (active sites, binding motifs, interfaces) that are often conserved beyond sequence similarity.
2 The key engineering idea is a compact inverted index over position-independent, pairwise geometric “feature encodings” derived from proximal residue pairs (<20 Å). Unlike prior motif indices that store positions, Folddisco stores only structure IDs (with delta compression), shrinking storage while keeping lookups fast.
3 Folddisco extends the classic RCSB-style residue-pair representation by adding two side-chain orientation features: dihedral angles (N1–CA1–CB1–CB2 and N2–CA2–CB2–CB1). These torsion features improve sensitivity and reduce an overly permissive prefilter (ablation reduces F1 and increases runtime).
4 Querying is a 4-stage pipeline: (i) extract/encode query pair features, (ii) prefilter candidates by index lookup (including tolerance-based “extended search”), (iii) residue matching via a compatibility graph and connected components, (iv) superposition and scoring (RMSD plus optional TM-score/GDT/Chamfer/Hausdorff).
5 Candidate ranking is driven by a rarity-aware coverage score: shared feature encodings are weighted by inverse document frequency (IDF), rewarding rare motif-defining geometry and down-weighting ubiquitous patterns (e.g., helix-like encodings). A length penalty helps avoid random matches in large proteins and keeps behavior consistent from tiny motifs to longer fragments.
6 Scale results: Folddisco builds an index for 53M AFDB50 structures in <25 hours with a 1.45 TB index, reported as ~11x faster to build and ~4x smaller than state-of-the-art pair-index approaches; querying is ~20x faster than pyScoMotif (and prefilter-only can be ~100x faster), with AFDB50 prefilter taking ~12 seconds.
7 Accuracy highlights: on human-proteome motif benchmarks, Folddisco notably improves recall for a full 4-residue C2H2 zinc-finger motif where RCSB/pyScoMotif show low sensitivity; for longer discontinuous segment queries, Folddisco outperforms MASTER while being much faster and using a much smaller index.
8 Generalization benchmarks: on SCOPe-derived “scattered conserved residue” queries (5,753 motifs), Folddisco shows substantially higher sensitivity than pyScoMotif (AUC 0.837/0.732/0.504 vs 0.285/0.290/0.300 for full/60%/20% conserved-column sampling). On M-CSA catalytic-site retrieval, Folddisco improves AUC over pyScoMotif (0.432 vs 0.344; a tuned “Sensitive” mode reaches 0.463).
9 Example applications shown: (i) motif-based functional annotation in sequence-divergent/uncharacterized proteins (including metagenomic and non-model-organism hits), (ii) distinguishing GPCR activation states using state-defining motifs (CWxP, NPxxY, DRY), (iii) cross-chain protein–protein interface motif search (immunoglobulin-like interfaces), plus demonstrations of double-motif querying, disulfide/knottin motif detection, and even short linear motif patterns.
10 Limitations discussed: the connected-component matching and 20 Å proximity constraint can miss widely separated multi-site motifs (e.g., distant allosteric pockets), fixed binning can lose borderline matches, and IDF coverage ranking is not always optimal for very short motifs (RMSD ranking can help there). The authors propose motif-specific E-values and variable binning, and aim to extend to nucleic-acid and ligand motifs.
💻Code: folddisco.foldseek.com; github.com/steineggerlab/fol…
📜Paper: nature.com/articles/s41587-0…
#computationalbiology #bioinformatics #structuralbiology #proteins #alphafold #motifsearch #proteinfunction #enzymes #GPCR #proteininterfaces
15
69
3,879

May 29
Excited to share our #RECOMB2026 work, DETANGO: a deep learning framework for disentangling mutation effects on protein stability and function from evolutionary signals captured by protein language models (pLMs).
DETANGO estimates a functional plausibility score that quantifies mutation effects on function beyond what can be explained by stability alone. Across extensive benchmarks, DETANGO accurately identifies stable-but-inactive (SBI) variants and functionally important residues involved in ligand binding, catalysis, and allostery.
Grateful to my co-authors @ZiangLi2001, @Qwe1029384756Tu, and Jiaqi Luo, and to my advisor @luoyunan for their invaluable contributions and support throughout this work! Special thanks to Tony for representing our team and presenting DETANGO today at RECOMB 2026 in Thessaloniki, Greece!
Preprint: biorxiv.org/content/10.64898…
Code: github.com/luo-group/DETANGO
#MutationEffectPrediction #ProteinLanguageModels #ProteinFunction #ComputationalBiology #DeepLearning
1
2
271
ProtSpace: Protein Universe in Your Browser
1 ProtSpace is a web-native viewer for protein language model (pLM) embedding spaces that runs entirely client-side, enabling interactive exploration of up to ~573K Swiss-Prot proteins in the browser with no installation, no login, and no data upload (privacy by design).
2 The core idea is to move beyond sequence-similarity networks (e.g., BLAST-derived graphs) and instead visualize learned embedding neighborhoods that can capture functional/structural relationships even when sequence similarity is weak, using interactive 2D projections as a hypothesis-generation interface.
3 A key engineering contribution is scale: WebGL-accelerated rendering plus a quadtree spatial index yields responsive hover/click/selection at hundreds-of-thousands of points; pan/zoom stays ~constant-time, while annotation switching and selection scale roughly linearly with dataset size in their benchmark suite.
4 The pipeline is designed for “bring your own proteins”: users can start from FASTA, a UniProt query, or precomputed HDF5 embeddings. Preparation is available via a zero-install Google Colab notebook or a Python CLI (protspace prepare), with caching and a YAML run log for reproducibility.
5 ProtSpace supports 12 embedding models via the Biocentral API (including ProtT5, multiple ESM-2 variants, Ankh, and ESM-C). For known proteins, UniProt’s precomputed ProtT5 embeddings can eliminate the need for local GPUs.
6 The annotation system is unusually rich for an embedding viewer: 38 switchable annotation types are automatically retrieved from five sources (UniProt, InterPro, NCBI Taxonomy, TED structural domains via AlphaFold DB, and Biocentral predictors), and can be augmented/overridden by user-provided CSV metadata.
7 It explicitly represents uncertainty/provenance: UniProt-derived evidence codes (ECO) are preserved and shown for key fields (e.g., GO aspects, EC numbers, subcellular localization, protein families), while InterPro domain hits expose per-domain scores and TED domains include pLDDT-based confidence.
8 A distinctive visualization feature is multi-label rendering as per-point pie charts (e.g., domain architectures such as multiple Pfam hits), allowing users to see compositional patterns across embedding neighborhoods rather than forcing single-label coloring.
9 ProtSpace integrates structure into the same exploratory loop: selecting a protein can open an embedded Mol* viewer that loads AlphaFold predictions via 3D-Beacons, enabling rapid cross-checks between embedding proximity, annotations, and structural context.
10 The paper’s examples emphasize multi-scale biological use: (i) Swiss-Prot-wide organization separates broad domains of life; (ii) joint human–Drosophila projections reveal overlap for conserved families and isolated regions enriched for lineage-specific families; (iii) beta-lactamase superfamily maps recover known Ambler-class structure and flag candidates for misannotation (e.g., proteins clustering with mechanistically different groups) and potential novel family clusters.
💻Code: github.com/tsenoner/protspac…
📜Paper: biorxiv.org/content/10.64898…
#Bioinformatics #ComputationalBiology #ProteinLanguageModels #Embeddings #ProteinFunction #UniProt #InterPro #AlphaFold #WebGL #Visualization

8
39
2,608

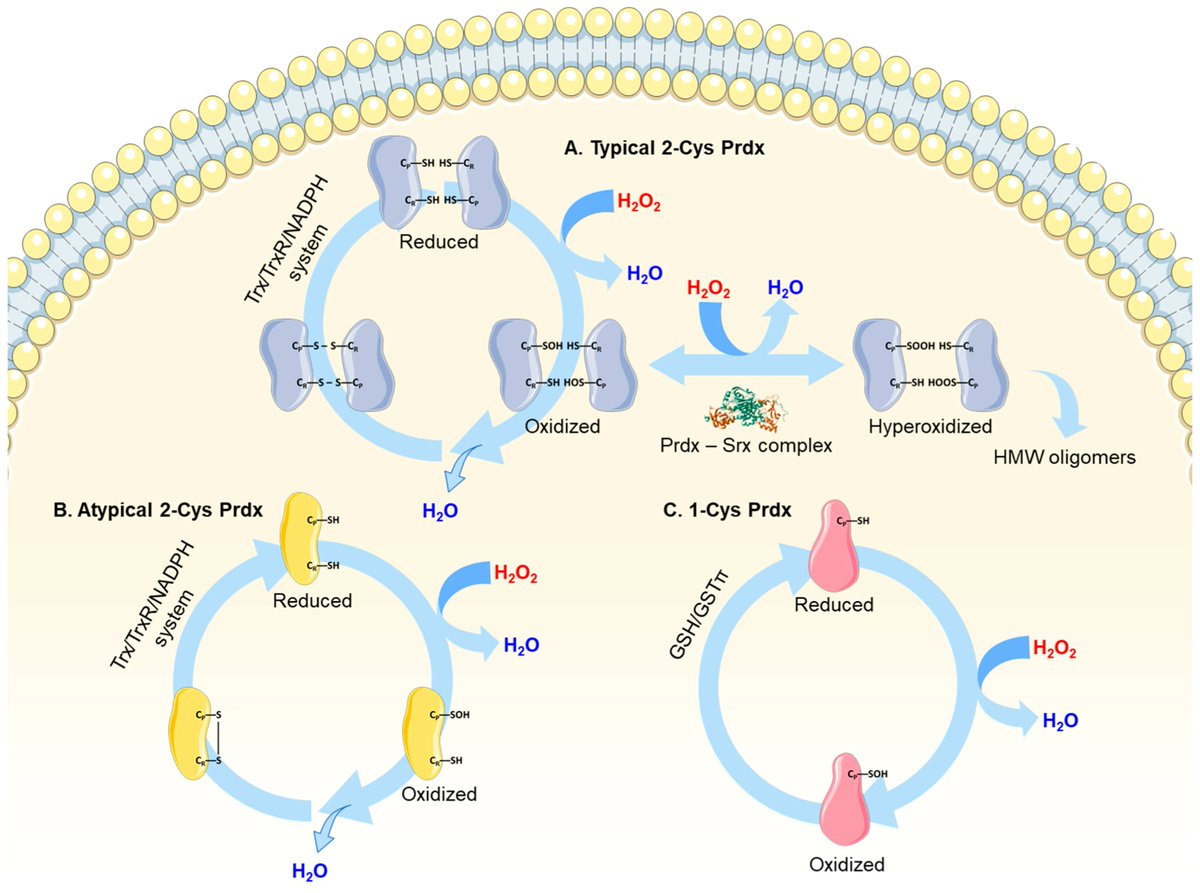
🔬 Are antioxidant enzymes doing more than just fighting oxidative stress?
📄 Moonlighting Functions of Mammalian Peroxiredoxins in Cellular Signaling
✍️ Yosup Kim et al.
🔗 brnw.ch/21x1XjK
#RedoxBiology #CellSignaling #Antioxidants #ProteinFunction #OxidativeStress
1
52

1/2: StarFunc: Fusing Template-based and Deep Learning Approaches for Accurate Protein Function Prediction
By Chengxin Zhang @ChengxinZhang3
#Bioinformatics #ProteinFunction #AI #CAFA5 #Genomics
academic.oup.com/gpb/advance…
1
1
145

Apr 6
Milyonlarca proteinin işlevi hala bilinmiyorken yapay zeka bu boşluğu kapatabilir mi?
Dünya genelinde 250 milyondan fazla protein dizisi veritabanlarında kayıtlı olmasına rağmen bunların sadece yüzde birinden azı deneysel olarak işlevsel açıdan tanımlanmış durumdadır. Geleneksel hesaplamalı yöntemler yüzeysel dizi benzerliğine dayanarak ya da her proteini bağımsız bir sınıflandırma problemi olarak ele almaktadır.
BioReason-Pro; protein dizisini, yapısını, evrimsel bağlamı ve etkileşim ağlarını bir arada değerlendirerek uzman biyologların akıl yürütme biçimini modellemeyi hedeflemiştir. GO-GPT adlı tamamlayıcı model ise gen ontolojisi terimlerini hiyerarşik ilişkileriyle birlikte otomatik bağlanımlı biçimde üretmektedir. 133.492 protein ve 3.135 organizma üzerinde yapılan değerlendirmede sistem mevcut yöntemlerin tamamını geride bırakmıştır.
📊 Çalışma, yapay zekanın hekimlerin ya da biyologların yerini almak yerine, işlevi bilinmeyen proteinlerin karakterizasyonunda güçlü bir akıl yürütme ve bilgi sentezleme aracı olabileceğini göstermiştir.
📙Dil Modelinden Biyolojik Akıl Yürütmeye: BioReason-Pro ile Protein İşlev Tahmini hakkındaki haber yazısının devamı ve diğer bilimsel içerikli yazılarımız için web sitemizi ziyaret edebilirsiniz.
🔗: bioinforange.com/bioinfonews…
#artificialintelligence #bioinformatics #proteinfunction #ainscience #bioinforange

2
108
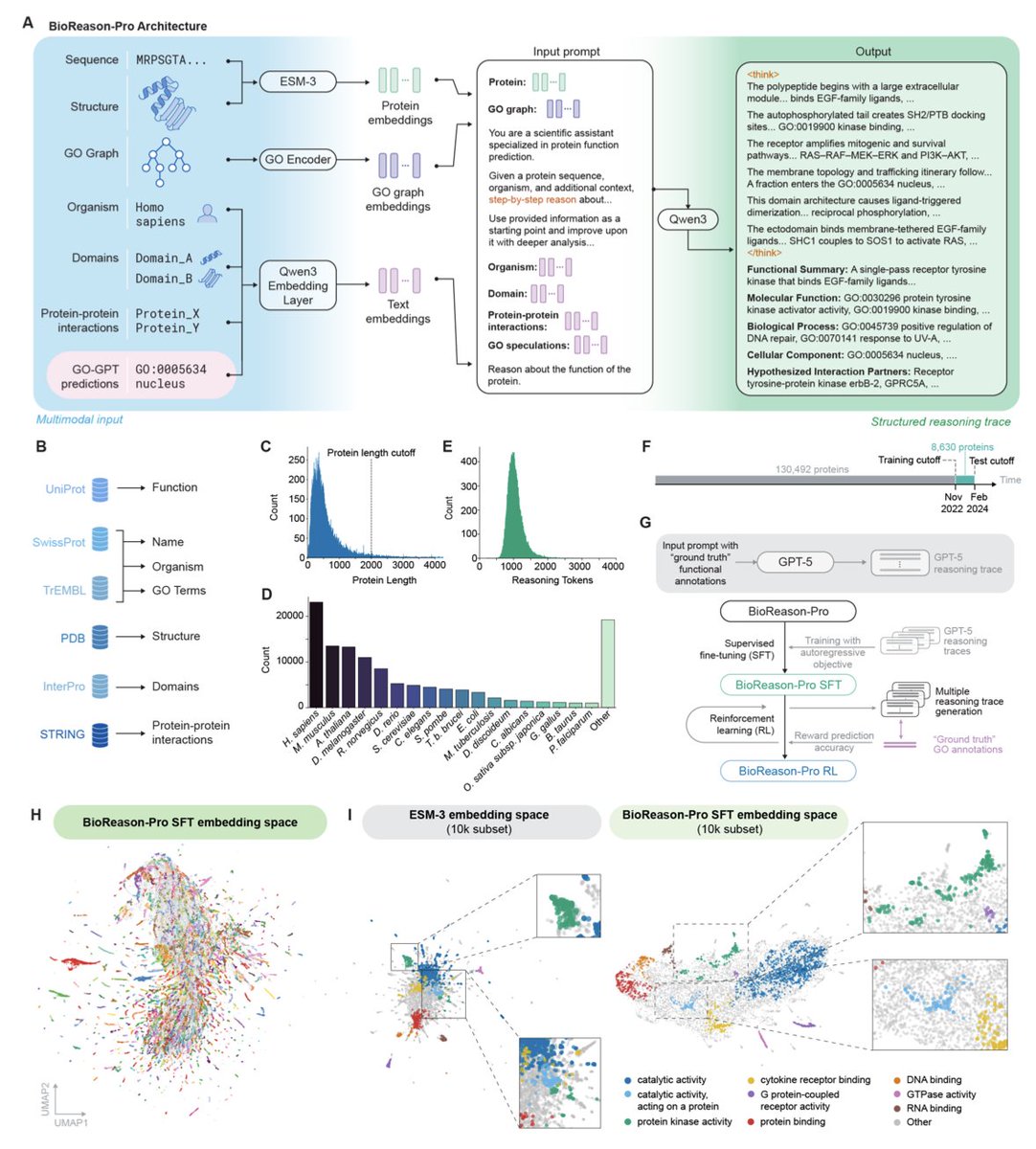
BioReason-Pro: Advancing Protein Function Prediction with Multimodal Biological Reasoning @arcinstitute
1. BioReason-Pro introduces the first multimodal reasoning large language model specifically designed for protein function prediction, combining protein embeddings with biological context to generate interpretable reasoning traces rather than just classification labels.
2. The system integrates ESM3 protein embeddings, a GO graph encoder, and biological context including organism, domains, protein-protein interactions, and GO-GPT predictions to perform step-by-step biological reasoning from sequence to function.
3. GO-GPT, a key component, is the first autoregressive transformer for Gene Ontology prediction that captures hierarchical and cross-aspect dependencies between GO terms, achieving state-of-the-art Fwmax of 0.65-0.70 across inference strategies.
4. The model was trained on over 130,000 synthetic reasoning traces generated by GPT-5 and further optimized through reinforcement learning with Group Sequence Policy Optimization, achieving 73.6% Fmax on GO term prediction.
5. Human protein experts preferred BioReason-Pro annotations over ground truth UniProt annotations in 79% of evaluated cases, with an LLM judge score of 8/10 for functional summaries, substantially outperforming previous methods.
6. Remarkably, BioReason-Pro de novo predicted experimentally confirmed binding partners with per-residue attention localizing to exact contact residues resolved in cryo-EM structures, demonstrating genuine structural reasoning capabilities.
7. The model successfully performed structural reasoning that overrode misleading superfamily-level domain annotations, such as correctly identifying CFAP61 as a non-enzymatic scaffold despite its Rossmann-like fold that typically indicates catalytic activity.
8. For eEFSec, BioReason-Pro identified SECIS-binding protein 2 as the obligate functional partner from sequence alone, with attention concentrated on the RIFT domain surface that matches the experimentally resolved SECIS RNA binding interface in PDB 7ZJW.
9. The system maintains strong performance even for proteins with very low sequence similarity to training data, with performance degrading much more slowly than BLAST as sequence identity decreases, indicating learned generalizable reasoning rather than simple homology transfer.
10. All model weights, code, and curated datasets are released publicly, alongside precomputed predictions for over 240,000 proteins including the Human Protein Atlas, enabling broad adoption for functional annotation of uncharacterized proteins.
💻Code: bioreason.net/code
📜Paper: biorxiv.org/content/10.64898…
#BioReasonPro #ProteinFunction #ComputationalBiology #Bioinformatics #MachineLearning #LLM #GeneOntology #ProteinStructure #FunctionalAnnotation #AIforScience
1
20
84
5,556
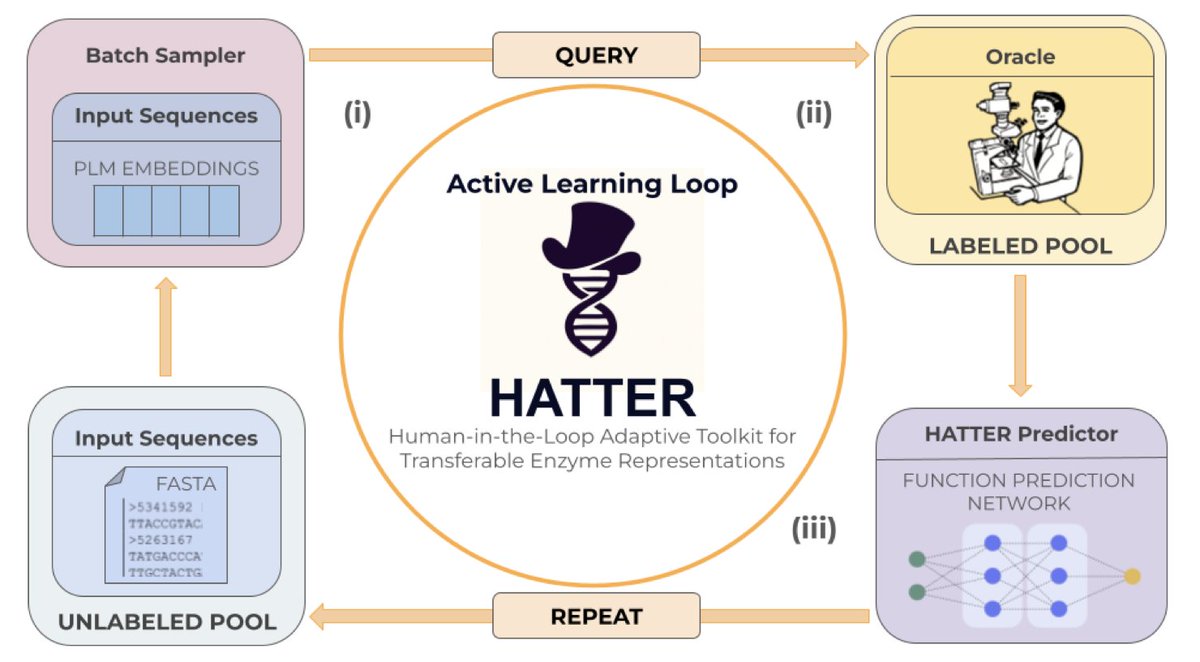
An Active Learning Framework for Data-Efficient, Human-in-the-Loop Enzyme Function Prediction
1. Introducing HATTER: a first-ever human-in-the-loop active learning framework specifically designed for enzyme function prediction, enabling biologists to iteratively annotate and update models alongside experimental workflows.
2. The framework achieves statistically comparable performance to standard supervised training while processing up to 48% less data and requiring fewer model updates, directly addressing the bottleneck between exponentially growing protein sequences and slow experimental validation.
3. Surprisingly simple point-based uncertainty methods like entropy and margin sampling outperform complex Bayesian approaches, highlighting that sequence diversity matters more than sophisticated acquisition functions for this biological task.
4. HATTER's modular design supports multiple architectures including CLEAN and custom neural networks, with four operational modes: training, initialization, update, and simulation for pre-experimental planning.
5. The system is explicitly built for real experimental constraints: adjustable batch sizes as small as 1, annotated query exports, and efficient model weight storage to avoid retraining between rounds.
6. Key insight for practitioners: random sampling performs surprisingly well, suggesting that capturing input diversity may be more important than optimizing uncertainty metrics when dealing with highly diverse biological sequences.
7. This work establishes a foundation for adaptive AI systems in biology that evolve with new data and expert input, moving beyond static benchmarks toward collaborative, iterative discovery platforms.
💻Code: github.com/ahoarfrost/HATTER
📜Paper: arxiv.org/abs/2602.23269
#ActiveLearning #EnzymeDiscovery #ProteinFunction #Bioinformatics #MachineLearning #ComputationalBiology #HumanInTheLoop #DataEfficiency
1
1
23
1,718

Finally, it investigates PAAC-based unsupervised clustering as an unbiased alternative, offering best-practice recommendations for accurate and interpretable computational annotation. #ComputationalBiology #Bioinformatics #ProteinAnnotation #ProteinFunction(4/4)
1
75
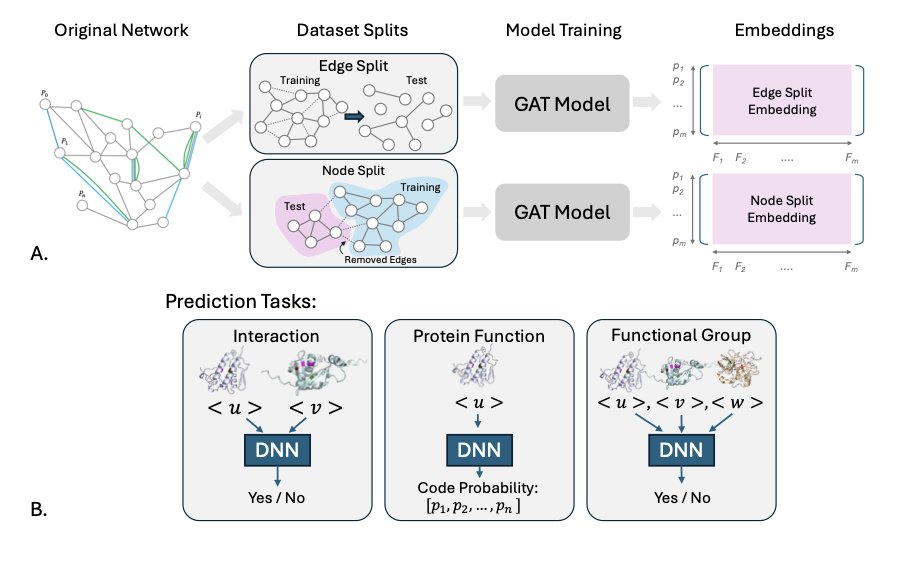
GATSBI: Improving context-aware protein embeddings through biologically motivated data splits
1 The most striking finding: evaluation strategy matters more than you might think. The authors show that standard random splits in protein embedding research massively overestimate real-world performance, especially for understudied proteins that actually need computational help.
2 GATSBI introduces task-aligned data splits that mirror real biological scenarios. Edge splits test recovering missing relationships among known proteins, while inductive node splits evaluate generalization to entirely unseen proteins—critical for annotating newly discovered genes.
3 The framework integrates heterogeneous biological networks (protein-protein interactions, co-expression, tissue-specific associations) with ESM-2 sequence embeddings using graph attention networks that learn context-aware representations.
4 Performance gains are largest where they matter most: understudied proteins show AUROC improvements of 0.259 over existing methods, compared to 0.244 for well-studied proteins. This reverses the usual bias toward proteins that already have abundant experimental data.
5 The node split evaluation reveals a key trade-off. While edge splits excel at interaction prediction (AUROC 0.88), node splits prove superior for functional annotation of novel proteins—demonstrating that no single benchmark captures all use cases.
6 Qualitative analysis suggests GATSBI's "false positives" may actually be unreported biological relationships. Predicted interactions between understudied proteins like Protocadherin-15 and Stereocilin align with evidence from model organisms.
7 The embedding space visualization shows understudied proteins cluster near well-studied neighbors with average cosine distance of 0.23, explaining how the model enables effective knowledge transfer from data-rich to data-poor regions of the proteome.
8 This work provides a sobering reminder: current benchmarks favor proteins that least need computational prediction. The authors release embeddings and code to enable more realistic evaluation in future protein representation learning research.
💻Code: github.com/Helix-Research-La…
📜Paper: biorxiv.org/content/10.64898…
#ProteinEmbeddings #GraphNeuralNetworks #ComputationalBiology #Bioinformatics #MachineLearning #ProteinFunction #UnderstudiedProteins #DataSplits #EvaluationMatters
3
8
1,118

Chemical proteomics links protein levels to function by mapping interactions. It reveals protein roles in complex biology 🔬🧬. #ChemicalProteomics #ProteinFunction #Interactome.
#Research #Medicine
doi.org/10.1039/d5cs00381d
30

Jan 5
Synapse: Your Connection to our MSK Authors
Meet: Richard Kevin Hite @HiteLabMSKCC
Research Focus: Structural Biology; Associate Member
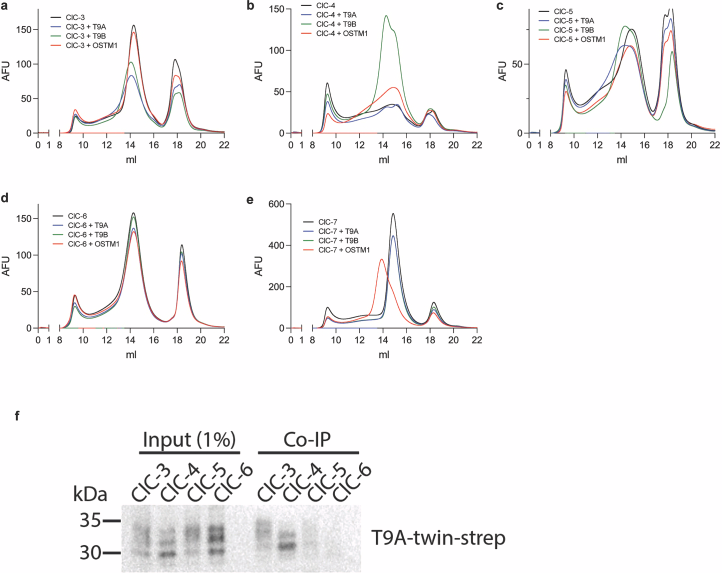
Structural basis of ClC-3 transporter inhibition by TMEM9 and PtdIns(3,5)P(2)
synapse.mskcc.org/synapse/wo…
#ScientificResearch #MolecularInsights
#BiomedicalResearch #ProteinFunction #CellSignaling

80

Jan 2
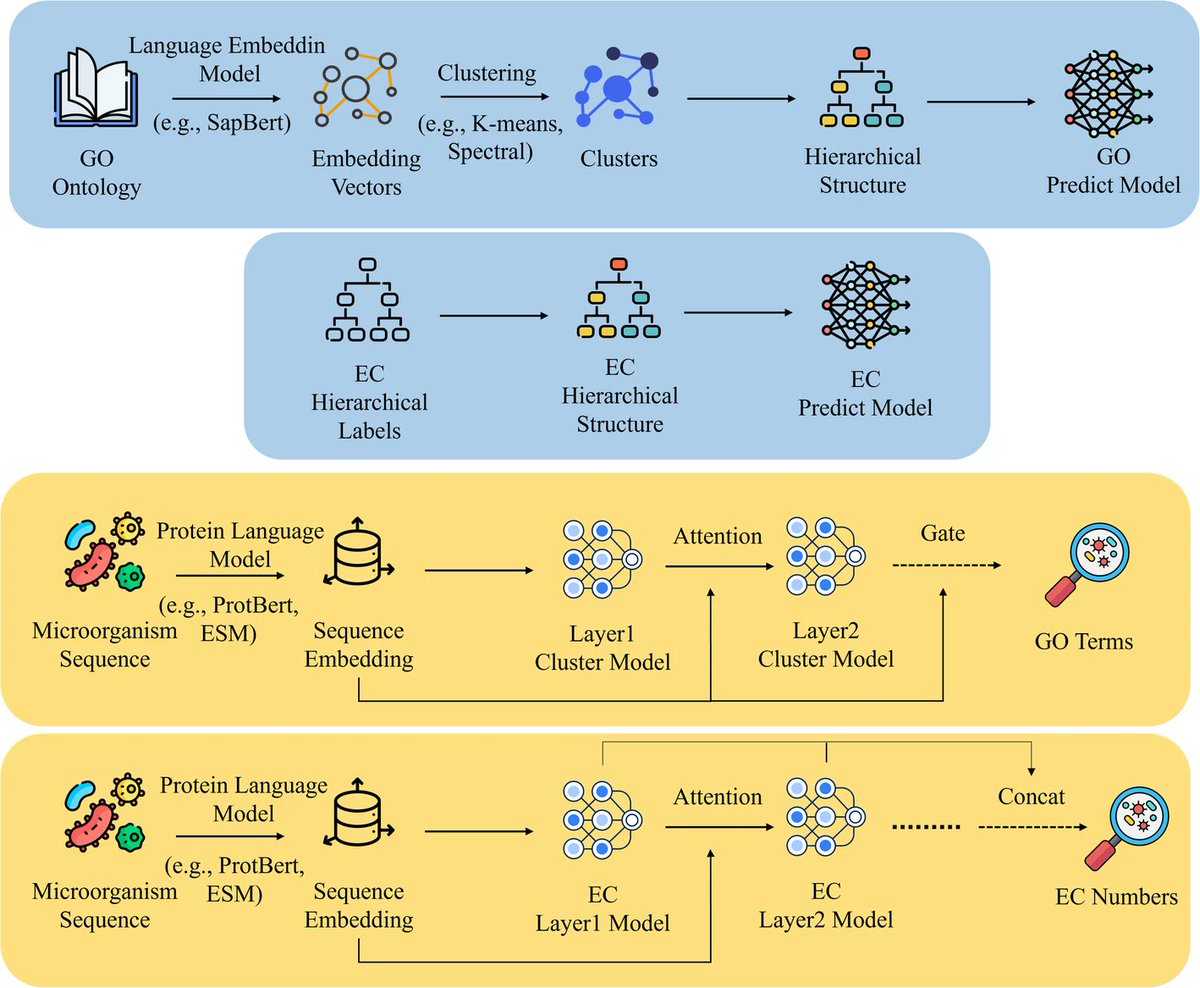
🔍 Predict microbial protein functions smarter! HCCN uses hierarchy attention-driven GO prediction for better accuracy. #MachineLearning #Bioinformatics #Microbiome #ProteinFunction #BMCBioInformaticsDetails: link.springer.com/article/10…
1
3
237

3 Dec 2025
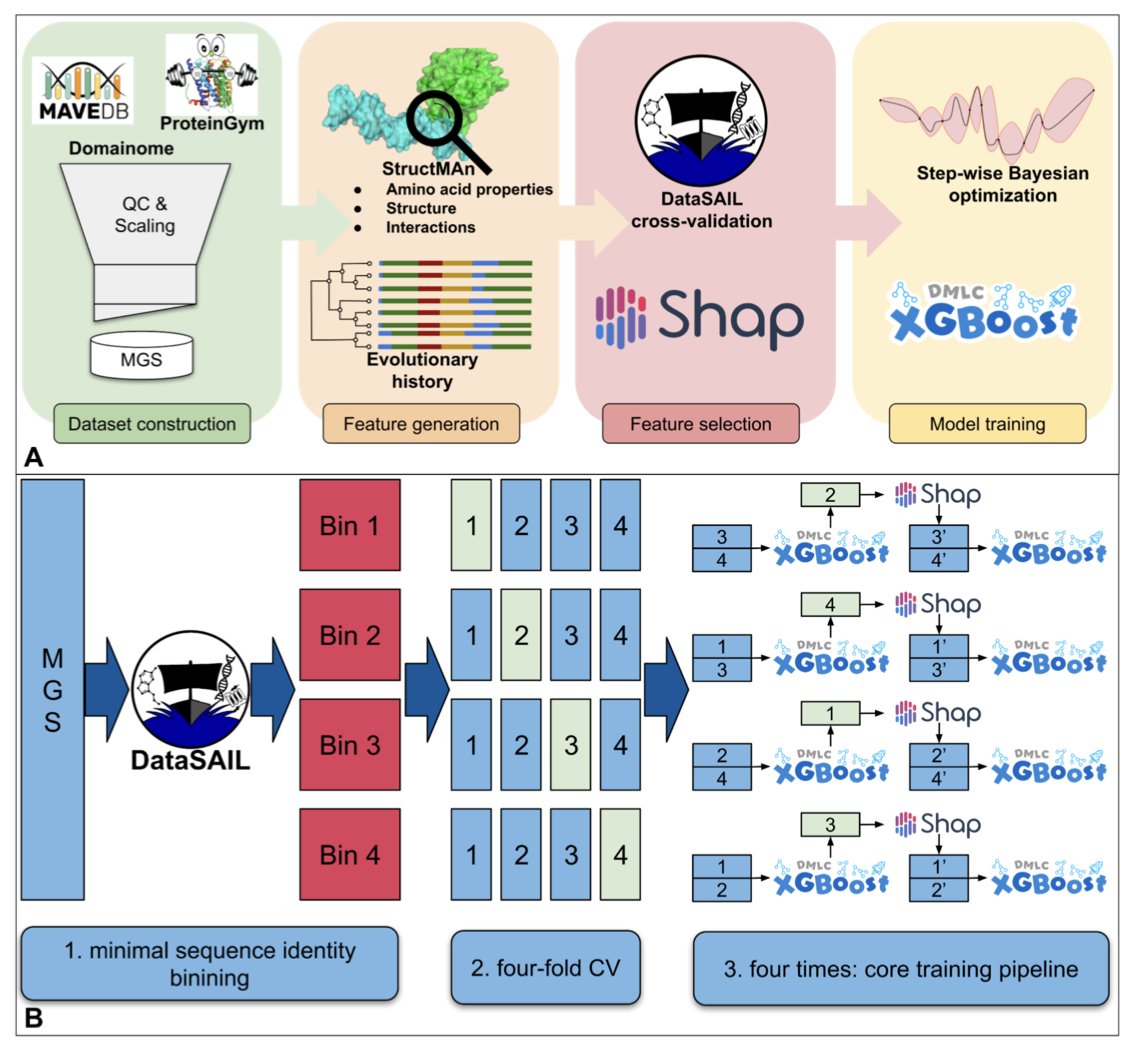
StructGuy: Data leakage free prediction of functional effects of genetic variants
1. A new supervised machine learning model, StructGuy, has been introduced to predict the functional effects of genetic variants on proteins. Unlike other models, StructGuy is designed to generalize predictions to proteins not seen during training, addressing a significant challenge in variant effect prediction.
2. The study leverages a dedicated training dataset derived from multiplexed assays of variant effects (MAVE) experiments. This dataset is carefully curated to prevent data leakage, ensuring that the model can accurately predict effects on unseen proteins.
3. StructGuy employs gradient boosting trees and integrates comprehensive protein structure-based features, including interactions and evolutionary information. This approach allows the model to provide fully interpretable predictions, offering insights into how mutations affect protein 3D structure.
4. The model's performance was evaluated using a modified ProteinGym benchmark, demonstrating competitive accuracy compared to state-of-the-art zero-shot methods. StructGuy's ability to generalize across different proteins makes it a valuable tool for variant effect prediction.
5. In a case study on the peroxisome proliferator-activated receptor gamma (PPARG), StructGuy successfully predicted the functional impact of mutations, illustrating its potential for mechanistic molecular hypotheses. This highlights the model's practical application in understanding variant effects at the protein level.
📜Paper: biorxiv.org/content/10.64898…
#StructGuy #VariantEffectPrediction #MachineLearning #ProteinFunction #Bioinformatics
3
11
1,321

3 Dec 2025
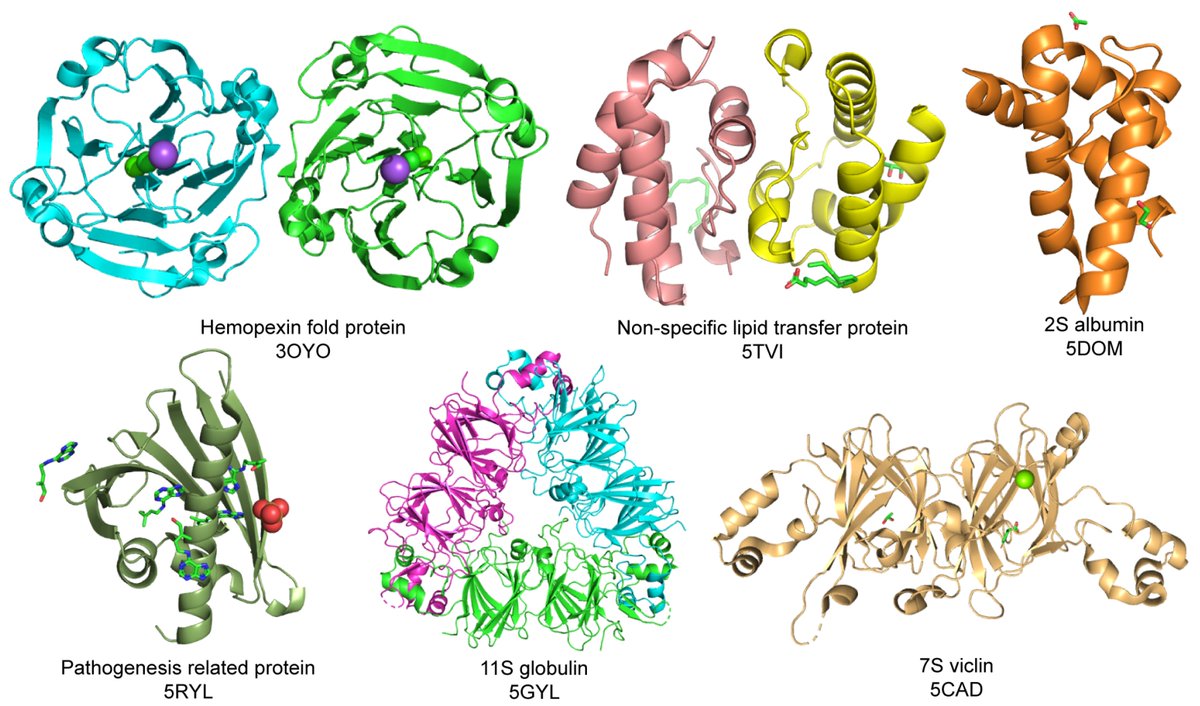
🔔 Welcome to read our highly cited paper published "Seed Storage Protein, Functional Diversity and Association with Allergy" by Dr Abha Jain et al.
🔗 Link: brnw.ch/21wY20A
#SeedProteins #PlantBiology #ProteinFunction #FoodAllergies #PlantScience #Protein
19
23 Nov 2025
TooTranslator: Zero-Shot Classification of Specific Substrates for Transport Proteins by Language Embedding Alignment of Proteins and Chemicals
1. Sima Ataei and Gregory Butler introduce TooTranslator, a novel model leveraging language embedding alignment to predict specific substrates for transmembrane transport proteins. This approach addresses the challenge of limited experimental data by using zero-shot learning, enabling predictions for substrates not seen during training.
2. The model integrates embeddings from ProtBERT, ChemBERTa, and SciBERT into a shared latent space, allowing substrate prediction through minimizing distances between protein and substrate embeddings. This multi-modal learning strategy is innovative in bridging protein sequences and chemical compounds.
3. TooTranslator demonstrates significant potential in zero-shot learning, achieving top-100 accuracy of 80% for unseen substrates. While direct predictions for unseen classes are limited, the model shows promise in aligning protein and chemical representations for broader applications.
4. The study highlights the importance of addressing class imbalance and data scarcity in transport protein classification. By using regression-based learning and various loss functions, TooTranslator provides a new perspective on open-world protein function prediction.
5. The dataset and code for this project are publicly available on GitHub, allowing researchers to explore and build upon this innovative approach. This work represents a valuable step towards improving our understanding of transport protein specificity.
📜Paper: biorxiv.org/content/10.1101/…
#ZeroShotLearning #ProteinFunction #Bioinformatics #ComputationalBiology #LanguageModels

5
1,091
20 Nov 2025
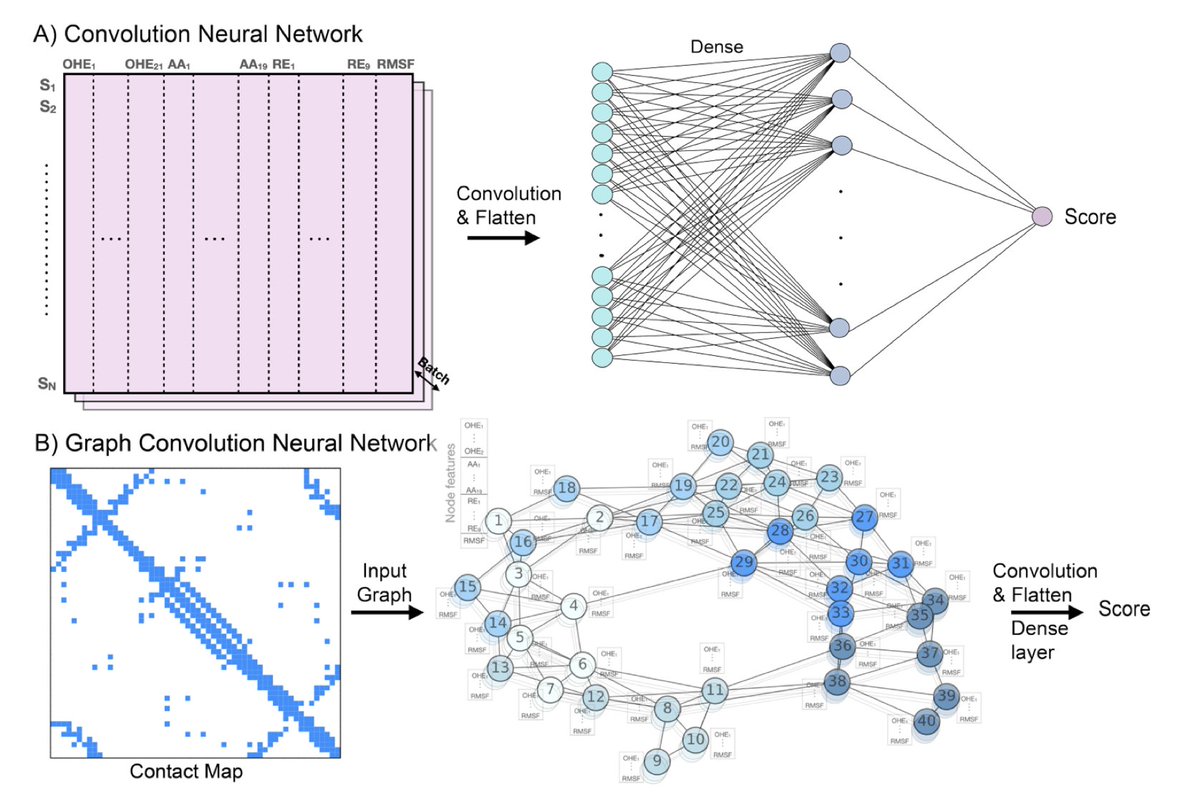
Overcoming Extrapolation Challenges of Deep Learning by Incorporating Physics in Protein Sequence-Function Modeling
1. This study introduces a novel approach to enhance deep learning models for predicting protein function from sequence data by integrating biophysical principles. The incorporation of physics-based features significantly improves the models' ability to extrapolate beyond training data, addressing a major limitation in current deep learning applications.
2. The authors demonstrate that using Rosetta-based energy terms and molecular dynamics simulations to quantify the effects of mutations can boost model performance. This method allows for more accurate predictions of protein function, especially in cases where training data is limited or incomplete.
3. The study evaluates the effectiveness of this approach on five diverse protein datasets, showing substantial improvements in positional and mutational extrapolation tasks. This suggests that leveraging biophysics can be a powerful strategy to overcome data scarcity and enhance model robustness.
4. The proposed method is efficient and scalable, requiring only residue-level energy evaluations. It can be applied to any protein or protein complex, making it a versatile tool for protein engineering and disease-related genetic studies.
5. The results highlight the importance of considering protein structure and dynamics in machine learning models. This work paves the way for future research combining advanced machine learning techniques with biophysical insights to further advance protein sequence-function modeling.
📜Paper: biorxiv.org/content/10.1101/…
#DeepLearning #ProteinEngineering #Biophysics #MachineLearning #ProteinFunction #ComputationalBiology
1
3
19
1,469

15 Nov 2025
The integration of genomics and natural language is fascinating! How does Genolator manage to handle the complexity of genomic and structural data together? It would be interesting to see how this model compares to existing genetic interpretation models in terms of accuracy and application. #AI #Genomics #ProteinFunction
8
15 Nov 2025
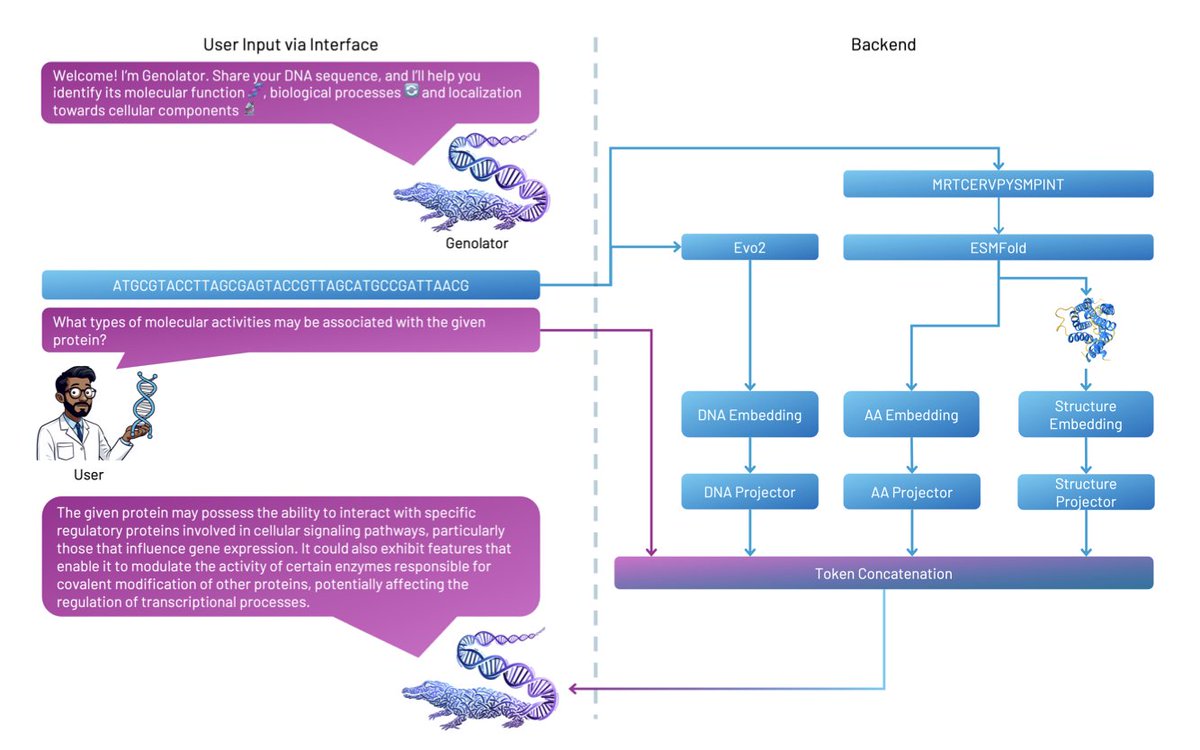
Genolator: A Multimodal Large Language Model Fusing Natural Language, Genomic, and Structural Tokens for Protein Function Interpretation
1. Genolator is a novel multimodal large language model that integrates natural language with genomic and protein structural data to interpret protein functions. This innovative approach bridges the gap between complex genomic information and human-readable insights, making it easier for researchers to explore and understand the functionality of proteins.
2. The model leverages embeddings from DNA sequences, amino acid sequences, and 3D protein structures, combining them with natural language queries. Fine-tuned on over 370,000 question-answer pairs, Genolator demonstrates high accuracy in confirming or denying protein function associations, outperforming baseline models like GPT-4 and specialized domain models.
3. A key innovation of Genolator is its ability to process multimodal data seamlessly. By using token projectors to fuse different types of embeddings, it enables a more comprehensive understanding of protein functionality. This multimodal integration allows the model to provide detailed answers to both open-ended and confirmatory questions about proteins.
4. Genolator's hidden states reveal a biologically and linguistically plausible organization of learned representations. The model clusters related Gene Ontology (GO) terms in a meaningful way, indicating its ability to capture the relationships between different biological processes, molecular functions, and cellular components.
5. The study also highlights the challenges of using general-purpose LLMs like GPT-4 for genomic tasks. Genolator shows that specialized multimodal models are more effective in handling complex genomic data, suggesting a promising direction for future research in computational biology.
6. The authors plan to release the code and trained models publicly, ensuring that the research community can reproduce and build upon this work. This openness will facilitate further advancements in using AI to decode the complexities of the human genome.
📜Paper: biorxiv.org/content/10.1101/…
#Genolator #MultimodalLLM #ProteinFunction #ComputationalBiology #Genomics #AIinBiology
1
7
29
2,180

19 Oct 2025
Solving the gene classification problem with a novel Gene Space approach
doi.org/10.1101/2025.10.17.6…
KIPEs3: Automatic annotation of biosynthesis pathways
doi.org/10.1101/2022.06.30.4…
#Bioinformatics #ProteinFunction
3
3
188