All-atomistic Transferable Neural Potentials for Protein Solvation
1 PHNN (Protein Hydration Neural Network) is introduced as an implicit solvent model that keeps the speed advantages of continuum solvation while learning environment-dependent corrections that remain predictive on out-of-domain proteins.
2 The key design shift vs typical “delta-learning” is that PHNN does not just add a post hoc energy correction; it learns corrections to the underlying GBn2 equation parameters (e.g., local dielectrics, screening, charges, SASA-related terms), so the analytical backbone remains the scaffold and the neural network fills known physical gaps.
3 PHNN is built on a GBn2 backbone and trained by force matching to explicit-solvent reference forces (CHARMM36 TIP3P), aiming to approximate mean solvation forces (PMF-consistent) using many instantaneous-force frames as noisy samples.
4 Architecture-wise, PHNN uses an E(3)-equivariant GNN (custom pseudo-MACE via cuEquivariance) to produce atom-centered embeddings that can represent higher-order geometric effects (up to quadrupolar information), which are relevant for anisotropic hydration structure and packing asymmetry near protein surfaces.
5 To avoid overfitting to stochastic instantaneous solvent forces, PHNN uses a heteroscedastic (variance-aware) training objective (β-NLL style). A separate invariant GNN estimates per-sample uncertainty using predicted forces plus key GBn2 parameters.
6 PHNN targets specific known GB/continuum failure modes with learned, physically interpretable modifications: (i) a learned modulation of the nonpolar SASA term with a learnable surface tension coefficient, (ii) atom-specific local solute dielectric and local solvent dielectric, (iii) a learned correction to the GB screening function to better handle mutual desolvation (important for salt bridges), and (iv) per-atom charge corrections to partially capture electrostriction-like effects.
7 On an independent OOD test (39 proteins), PHNN reports mean force MAE 66.6 ± 9.4 kJ/(mol·nm) vs explicit solvent, improving over GBn2 at 97.5 ± 9.0 kJ/(mol·nm) (about 31.7% lower error). The paper notes an intrinsic ceiling because explicit-solvent instantaneous forces have large variance, so deterministic implicit models cannot match every fluctuation.
8 In dynamical tests (4 domains, up to ~5400 atoms), PHNN better preserves native-like behavior than GBn2 when comparing RMSD/ROG/RMSF distributions and KDE-derived free-energy landscapes; GBn2 shows stronger unfolding tendencies, especially for larger domains.
9 Targeted error breakdowns suggest PHNN improves across secondary-structure classes and residue types; the largest gains are reported for lysine (consistent with improved salt-bridge screening). Remaining challenges include arginine (delocalized guanidinium charge is difficult to fix with per-atom corrections) and buried regions where long-range electrostatics may require larger interaction radii or deeper models.
10 Transferability limits are probed with alanine dipeptide (near-zero sequence similarity to training domains): PHNN reproduces major Ramachandran basins but distorts basin shapes and mis-ranks some regions (notably αR), motivating future training that explicitly enriches boundary/strained conformations via umbrella sampling and broader conformational coverage (including IDPs and 300K data).
📜Paper: arxiv.org/abs/2605.14584
#ComputationalBiophysics #MolecularDynamics #ImplicitSolvent #ForceFields #NeuralPotentials #EquivariantGNN #ProteinSimulation #Solvation #MLforScience

2
12
1,793

Quantum computing just scaled to 12,000-atom protein simulations. Here’s why it matters.
bit.ly/3R3wixd
#QuantumComputing #ProteinSimulation #QuantumChemistry

2
12
29
2,333
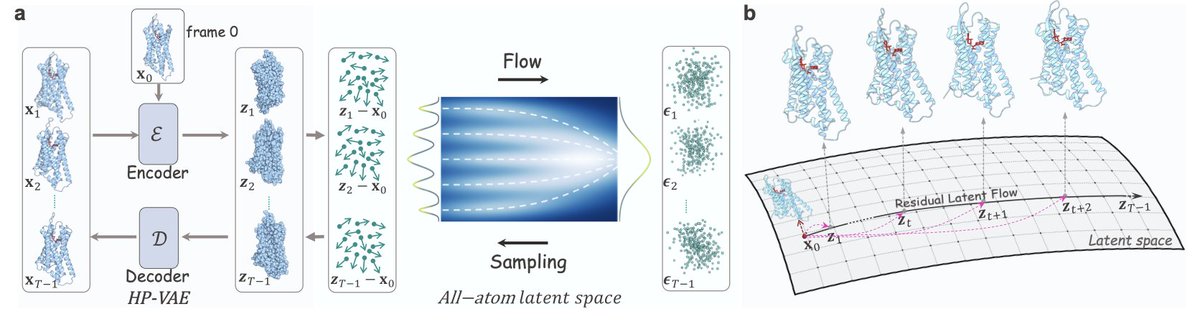
All-Atom GPCR-Ligand Simulation via Residual Isometric Latent Flow
1. GPCRLMD achieves a 900× speedup over traditional MD simulations, generating 100-500ns trajectories in seconds rather than days, making high-throughput GPCR drug screening computationally tractable for the first time.
2. The method introduces a Harmonic-Prior VAE that replaces the standard Gaussian prior with an atom-centered harmonic prior, preserving geometric topology by anchoring latent codes to their physical atomic positions rather than collapsing toward a featureless origin.
3. A Residual Latent Flow mechanism decouples static equilibrium structure from dynamic fluctuations by learning relative displacements from the initial frame, enabling stable generation of temporally coherent trajectories without drifting into unphysical configurations.
4. Unlike prior protein-only generative models, GPCRLMD explicitly models the coupled receptor-ligand dynamics, capturing ligand-induced conformational changes that are invisible to "ligand-blind" baselines like BioEmu.
5. The isometric latent space maintains full 3N atomic dimensionality rather than compressing into low-dimensional bottlenecks, preserving complete spatial resolution while still enabling efficient flow-based sampling on a regularized manifold.
6. Comprehensive benchmarking on 54 test GPCR-ligand complexes demonstrates state-of-the-art fidelity in reproducing RMSF profiles, contact maps, TM helix motions, and free energy landscapes compared to ground-truth MD simulations.
7. The architecture incorporates GPCR-specific domain knowledge including transmembrane helix topology and ligand pharmacological classification, though the framework is designed to generalize to broader protein-ligand systems.
📜Paper: arxiv.org/abs/2602.03902
#GPCR #MolecularDynamics #GenerativeAI #DrugDiscovery #ComputationalBiology #ProteinSimulation #FlowMatching #VariationalAutoencoder
2
13
56
3,346
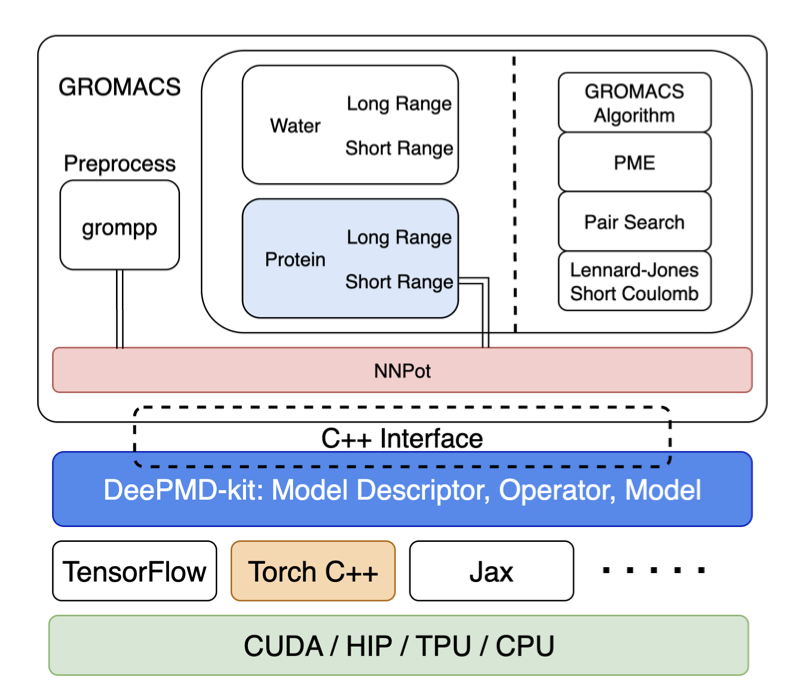
Enabling AI Deep Potentials for Ab Initio-quality Molecular Dynamics Simulations in GROMACS
1) This work brings AI deep potentials into GROMACS, a production-level molecular dynamics code, enabling ab initio-quality simulations at a fraction of DFT's computational cost through integration with DeePMD-kit.
2) The authors enable multiple deep potential model families and ML backends by coupling GROMACS Neural Network Potentials with DeePMD-kit's C /CUDA backend, providing a stable, forward-compatible path for quantum-mechanical forces in production MD pipelines.
3) Two recent large-atom-model architectures are evaluated: DPA2 (attention-based) and DPA3 (GNN-based), with DPA2 delivering up to 4.23× higher throughput on A100 and 3.18× on GH200 compared to DPA3.
4) The study reveals critical technical challenges in tight coupling: expanded ghost atom requirements for DPA models, asymmetric ghost topology handling, and the need for parallel inference support, all addressed through careful interface design.
5) GPU characterization shows DPA2 is GEMM-heavy (44% runtime) while DPA3 is memory-bound with indexing operations dominating (75% runtime), with DPA3 requiring 3× more GPU memory and hitting OOM limits on larger systems.
6) The linear scaling of deep potentials with atom count contrasts sharply with DFT's cubic scaling, though memory demands remain the primary bottleneck for large-scale biomolecular simulations.
7) Four solvated protein benchmarks (1YRF, 1UBQ, 3LZM, 2PTC) validate correct force evaluations and stable trajectories, demonstrating cross-system generalization of large atom models through downstream task specialization.
💻Code: github.com/HuXioAn/gromacs/t…
📜Paper: arxiv.org/abs/2602.02234
#MolecularDynamics #DeepLearning #ComputationalChemistry #GROMACS #DeePMD #AI4Science #HPC #GPUComputing #AbInitio #ProteinSimulation
2
3
31
1,656

22 Dec 2025
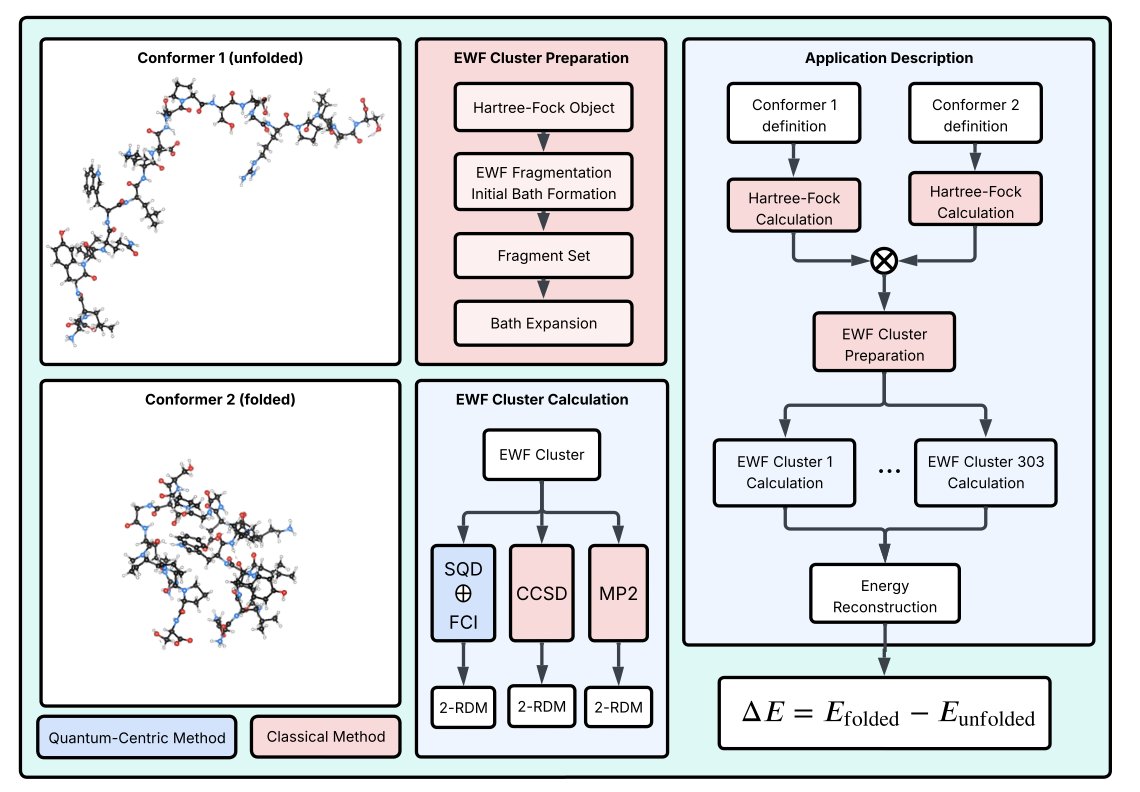
Molecular Quantum Computations on a Protein
1. A novel study presents the first application of quantum-centric supercomputing to simulate the electronic structure of a protein, specifically the Trp-cage miniprotein. This work pushes the boundary of quantum chemistry simulations to a new scale, handling systems with hundreds of atoms.
2. The research employs a fragment-based approach with wave function–based embedding (EWF) as the framework. It integrates sample-based quantum diagonalization (SQD) for challenging fragments and full configuration interaction (FCI) for simpler ones, showcasing a hybrid quantum-classical workflow.
3. A key innovation is the use of quantum hardware to sample electron configurations within the SQD method, combined with classical post-processing for error mitigation and energy calculations. This hybrid approach allows for simulations that are impractical with classical computing alone.
4. The study demonstrates that the SQD method can accurately approximate the ground-state energy of large protein fragments, with results compared against classical benchmarks like MP2 and CCSD. This highlights the potential for quantum computing to tackle complex biological systems.
5. The Trp-cage miniprotein serves as an ideal benchmark due to its compact size and distinct conformers. The workflow successfully predicts the relative energies of folded and unfolded states, illustrating the method's applicability to biologically relevant problems.
6. The research paves the way for future quantum simulations of larger biomolecular systems, potentially enabling detailed studies of protein dynamics, drug interactions, and other complex biochemical processes with unprecedented accuracy.
📜Paper: arxiv.org/abs/2512.17130v1
#QuantumComputing #QuantumChemistry #ProteinSimulation #HybridComputing #Bioinformatics
1
3
36
2,280
23 Sep 2025
Development of Shell-Space Electrostatic Potential Fitting Charges in a United-Atom Model for Amino Acids Simulations
1. A new method called Shell-Space Electrostatic Potential (SS-ESP) has been developed to accurately derive partial charges for coarse-grained (CG) particles in amino acid simulations. This method fits the electrostatic potential (ESP) of all-atom (AA) models to enhance the accuracy of CG models.
2. The SS-ESP method eliminates infinite values during the integration process, which is a significant improvement over previous methods. It uses a predefined shell space for numerical integration, ensuring consistent and accurate results.
3. The study proposes a new Side-chain Reduced CG (SRCG) model for amino acids, parameterized using a bottom-up strategy. The bonding interactions are inherited from the AA model, while the CG charges are derived using the SS-ESP method and the Lennard-Jones parameters are fitted to reproduce van der Waals interactions.
4. Molecular dynamics simulations show that the SRCG model accurately describes the solvation free energy of amino acids and preserves the stability of protein secondary structures. This makes it a promising tool for bottom-up development of protein CG models.
5. The SS-ESP charge fitting method provides a new framework for accurately describing the electrostatic interactions of CG particles. It outperforms other methods in terms of relative root-mean-square errors (RRMSEs) for ESPs and mean average errors (MAEs) for electric dipole moments.
📜Paper: doi.org/10.26434/chemrxiv-20…
#CoarseGrainedModeling #ElectrostaticPotential #AminoAcids #MolecularDynamics #ProteinSimulation

1
3
10
1,258

10 Jul 2025
🇩🇪 Germany | Funded PhD or Postdoc in Computational Biophysics – International Students Eligible
📍 Max Planck Institute for Multidisciplinary Sciences, Göttingen
🧪 Dept. of Theoretical & Computational Biophysics
👨🏫 Supervisor: Prof. Dr. Helmut Grubmuller
🔗 More: phdscanner.com/phd-vacancies…
Exciting opportunity to explore non-equilibrium theory and atomistic simulations of complex biomolecules. Possible research areas include:
Variational free energy methods
Constant-pH simulations
Thermodynamics of solvation shells
Functional mechanisms (ribosome, CRISPR/Cas, IDPs & more)
🎓 PhD applicants: Master’s in physics, mathematics, or related fields
🎓 Postdoc applicants: PhD in a relevant discipline
💡 Strong skills in scientific computing & statistical mechanics are essential
🖥️ Access to advanced GPU/high-performance computing
🗣️ English-speaking lab (no German required)
💰 Funded for 3 years (PhD) | 2 years (Postdoc, extendable)
📅 Rolling applications | Flexible start date
📩 Apply with cover letter, CV, and transcripts (English or German)
📧 Email: ausschreibung10-25@mpinat.mpg.de
#PhD #Postdoc #Biophysics #ComputationalBiology #MaxPlanck #GPUComputing #StructuralBiology #AcademicJobs #ResearchOpportunity #GermanyJobs #ProteinSimulation #CRISPR #PhysicsJobs #PhDAbroad #PostdocAbroad #ScienceJobs #InternationalStudents #STEMCareers
1
3
521

17 Aug 2023
Innovative approach simulates large-scale conformational change in biological systems using novel barrier restraints & 3-stage procedure for realistic path collective variable generation, advancing conformationally-driven drug discovery #ProteinSimulation
go.acs.org/5RV
2
4
1,196

30 May 2022
Computer: Rechnen durchbricht Exaflop-Grenze. Proteinsimulation erreicht erstmals eine Trillion Gleitkommaberechnungen pro Sekunde. #Exaflop #Supercomputer #Exascale #Rechentempo #Computer scinexx.de/news/technik/comp…
2
4


12 Nov 2016
Join the @BSC_CNS as web application software developer. Deadline: 30/11 bioinformaticsbarcelona.eu/j… #proteinsimulation #job #SoftDev
30 Oct 2016
Join the @BSC_CNS as web application software developer. Deadline: 30/11 bioinformaticsbarcelona.eu/j… #proteinsimulation #job #SoftDev
2
1
27 Oct 2016
Jr web application software development position - Life Sciences Dept @BSC_CNS Deadline: 30/11 bioinformaticsbarcelona.eu/j… #proteinsimulation
19 Oct 2016
Junior web application software development postion at the Life Sciences Department @BSC_CNS bioinformaticsbarcelona.eu/j… #proteinsimulation
7