
9/25 𝗖𝗼𝗺𝗽𝘂𝘁𝗮𝘁𝗶𝗼𝗻-𝗔𝘄𝗮𝗿𝗲 𝗞𝗮𝗹𝗺𝗮𝗻 𝗙𝗶𝗹𝘁𝗲𝗿𝗶𝗻𝗴 𝘄𝗶𝘁𝗵 𝗠𝗼𝗱𝗲𝗹 𝗦𝗲𝗹𝗲𝗰𝘁𝗶𝗼𝗻 𝗳𝗼𝗿 𝗡𝗲𝘂𝗿𝗮𝗹 𝗗𝘆𝗻𝗮𝗺𝗶𝗰𝘀
This paper introduces the Computation-Aware State-Space Model (CASSM), a framework for dynamical latent variable modeling of single-cell neural recordings, specifically for the scale-imbalanced regime (trials significantly lower than neurons). CASSM extends computational uncertainty to model selection using a novel training loss and optimization scheme, achieving tractable inference in large state-spaces. It demonstrates competitive performance with data-hungry deep networks and significantly improved uncertainty calibration on both synthetic and real data, offering a roadmap for neuroscience researchers.
#CASSM #Neuroscience #BayesianMethods #StateSpaceModels #UncertaintyQuantification #DynamicalLatentModels
Paper Link: arxiv.org/abs/2606.01468

1
23

May 21
Gated DeltaNet-2 is here. 🚀
🔥 New paper: Gated DeltaNet-2: Decoupling Erase and Write in Linear Attention
Gated DeltaNet-2 outperforms KDA and Mamba-3, the latest and best recurrent architectures, head to head at 1.3B. 🏆
💡 Here's the idea behind it:
Linear attention squeezes an unbounded KV cache into a fixed-size recurrent state. The hard part isn't just what to forget, it's how to edit that memory without scrambling the associations already in it.
Prior delta-rule models like Gated DeltaNet and KDA use one scalar gate to do two jobs at once: erasing old content and writing new content. But these two decisions act on different axes of the state, so tying them together is a real limitation.
Gated DeltaNet-2 decouples them.
✂️ a channel-wise erase gate b_t picks which key-side coordinates to read and remove
✍️ a channel-wise write gate w_t picks which value-side coordinates to commit
🔁 recovers KDA when both gates collapse to a scalar, and Gated DeltaNet when the decay collapses too
⚡ still trains fast: chunkwise WY algorithm with gate-aware backward, fused in Triton
📊 Results:
We train 1.3B models on 100B tokens of FineWeb-Edu, matched in recurrent state size, against Mamba-2, Gated DeltaNet, KDA, and Mamba-3.
Best average on language modeling commonsense reasoning, in both recurrent and hybrid settings
Biggest gains on long-context RULER retrieval. S-NIAH-3 jumps from 63 to 90 over KDA, and multi-key needle retrieval climbs from 28 to 38
Joint work with @YejinChoinka and @jankautz.
📄 Paper: shorturl.at/AAlVb
💻 Code: github.com/NVlabs/GatedDelta…
#LinearAttention #StateSpaceModels #Mamba #LLM

25
98
653
194,070
EMReady2: Improvement of cryo-EM and cryo-ET maps by local quality-aware deep learning with Mamba
1. EMReady2 is a real-space, single-map post-processing method that targets a key pain point in cryo-EM/cryo-ET interpretation: strong local heterogeneity (variable local resolution, anisotropy, fragmented connectivity) that makes a “one-size-fits-all” sharpening strategy unreliable.
2. The main architectural shift is BiMCUnet, a dual-branch UNet that fuses a bidirectional Mamba (state-space) branch for long-range/global context with a convolutional branch for local detail, aiming to keep global coherence while restoring local connectivity—without the quadratic cost of attention.
3. A core training innovation is local quality-aware supervision: instead of generating targets with a uniform resolution, EMReady2 builds simulated maps using atom-wise local resolution estimated from Q-scores, then blends local and global resolution (w=0.8). This is designed to reduce bias from poorly modeled regions and better match real map heterogeneity.
4. The training set is substantially broadened versus the original EMReady: balanced inclusion of proteins and nucleic acids, plus medium/low-resolution cases (6–10 Å) and cryo-ET subtomogram averaging (STA) maps—expanding the practical scope to 2–10 Å and to RNA/DNA-heavy assemblies.
5. On 118 diverse cryo-EM SPA maps (2–10 Å), EMReady2 improves map–model FSC-0.5 for 113/118 cases and leads across multiple metrics (mean FSC-0.5 4.65 Å vs 5.78 Å deposited; Q-score 0.493 vs 0.454; main-chain Q-score 0.557 vs 0.491), outperforming DeepEMhancer, phenix.auto_sharpen, EMReady, and CryoTEN in the reported benchmarks.
6. For nucleic-acid–rich maps (>10% nucleic acids; 18 cases), EMReady2 shows larger gains, consistent with the expanded training distribution (mean FSC-0.5 4.20 Å vs 5.70 Å deposited; Q-score 0.499 vs 0.453; main-chain Q-score 0.543 vs 0.462). Segmented evaluation suggests improvements are even stronger within nucleic-acid regions than globally.
7. EMReady2 extends to cryo-ET STA: on 18 STA maps (3–10 Å), it improves masked FSC-0.5, Q-score, CC_mask, and main-chain Q-score versus deposited maps and competing post-processing tools, supporting use beyond SPA where maps are typically lower resolution and noisier.
8. Interpretability is evaluated via automated de novo model building (phenix.map_to_model) for maps better than 5 Å: across 832 chains, models built from EMReady2-processed maps show higher residue coverage and sequence recall for both proteins (coverage 70.51%, recall 32.24%) and nucleic acids (coverage 72.14%, recall 27.40%) than deposited maps and other post-processing methods.
9. The paper explicitly probes “hallucination” risk on intrinsically disordered regions and reports that EMReady2 improves contrast/connectivity without forcing ambiguous density into canonical helices/sheets; it also emphasizes that conformational/compositional heterogeneity must be handled upstream, and that small ligands/ions/waters remain challenging for data-driven enhancement.
10. Efficiency is a practical highlight: average runtime across 136 maps is reported as 14.81 s, faster than CryoTEN (17.33 s), and far faster than EMReady (83.26 s) and DeepEMhancer (90.98 s), aligning with the linear-scaling motivation for adopting Mamba-style state-space modeling.
💻Code: github.com/huang-laboratory/…
📜Paper: doi.org/10.1038/s41467-026-7…
#cryoEM #cryoET #structuralbiology #computationalbiology #deeplearning #Mamba #statespacemodels #UNet #mapsharpening #nucleicacids
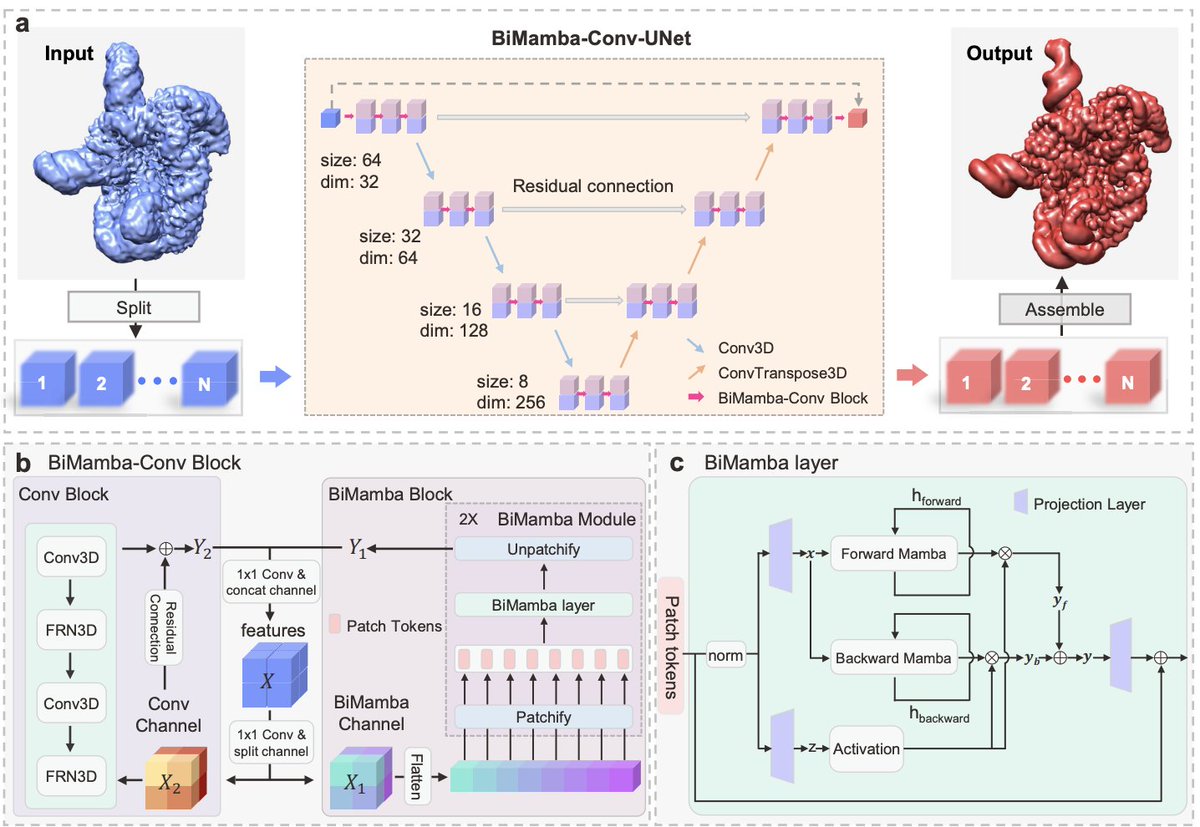
4
31
2,759
RegFormer: A single-cell foundation model powered by gene regulatory hierarchies @NatureComms
1. RegFormer is a single-cell foundation model that bakes gene regulatory hierarchies directly into the input representation: genes are ordered by a GRN-derived directed acyclic graph (DAG), so upstream regulators precede downstream targets during sequence modeling.
2. The architecture replaces Transformer attention with Mamba state-space blocks (linear-time in sequence length), enabling long-context modeling up to 10,000 genes and addressing the scalability/context-length limits common in attention-based single-cell foundation models.
3. Each gene is encoded with dual embeddings: a token embedding for gene/regulatory identity plus a value embedding for continuous expression magnitude; ablations show removing token embeddings or binarizing expression prediction degrades clustering quality, supporting the need for both regulatory context and quantitative modeling.
4. Pretraining combines three self-supervised objectives with fixed weights: masked value prediction (MVP), gene expression prediction for cells (GEPC), and regulatory topology/role prediction (TOPO). Objective ablations show the largest drop when TOPO is removed, highlighting the contribution of explicitly learning regulatory directionality.
5. Training data scale: 25 million human scRNA-seq profiles across 45 tissues and diverse technologies/conditions, harmonized via a unified pipeline and stored in LMDB for efficient streaming during large-scale pretraining.
6. GRN construction: TF–target links come from CisTarget motif-to-gene rankings (top-20 targets per TF), refined with Node2Vec embeddings and iterative edge pruning to remove cycles while retaining >92% of edges and preserving key graph statistics; genes are then topologically sorted for model input.
7. Benchmarks (BioLLM framework): across multiple tissue datasets, RegFormer yields higher Average Silhouette Width (ASW) for cell-type separation and improved batch integration compared with scGPT, Geneformer, scFoundation, and scBERT; the 10k-gene variant generally improves resolution by providing broader regulatory context.
8. Cell type annotation: RegFormer achieves higher Macro-F1 across diverse datasets, shows strong performance on fine-grained immune labels (e.g., Zheng68k), and improves rare cell type recognition; even a frozen backbone transfers well, with finetuning providing additional gains.
9. Mechanistic/biological fidelity: gene embeddings better align with GRN topology (e.g., TF vs non-TF classification; correlations with regulatory degree). GRNs reconstructed from embedding cosine similarity show higher functional coherence (GO enrichment and functional similarity), with case studies recovering canonical regulons (e.g., GATA3 in T cells; SPI1/PU.1 in myeloid cells).
10. Generalization to interventions and pharmacology: when plugged into GEARS, RegFormer embeddings support accurate CRISPR perturbation response prediction (high agreement between predicted and observed Δ-expression; responsive genes lie closer in embedding space to perturbed TFs). Integrated into DeepCDR-style pipelines, RegFormer improves IC50 prediction (PCC/SRCC) including leave-drug-out tests, and enriches pathway-level interpretability (e.g., NF-κB with Bortezomib; p53-related signals with SN-38).
💻Code: github.com/BGIResearch/RegFo…
📜Paper: doi.org/10.1038/s41467-026-7…
#scRNAseq #SingleCell #FoundationModels #GeneRegulatoryNetworks #Mamba #StateSpaceModels #Bioinformatics #ComputationalBiology #DrugResponse #CRISPR
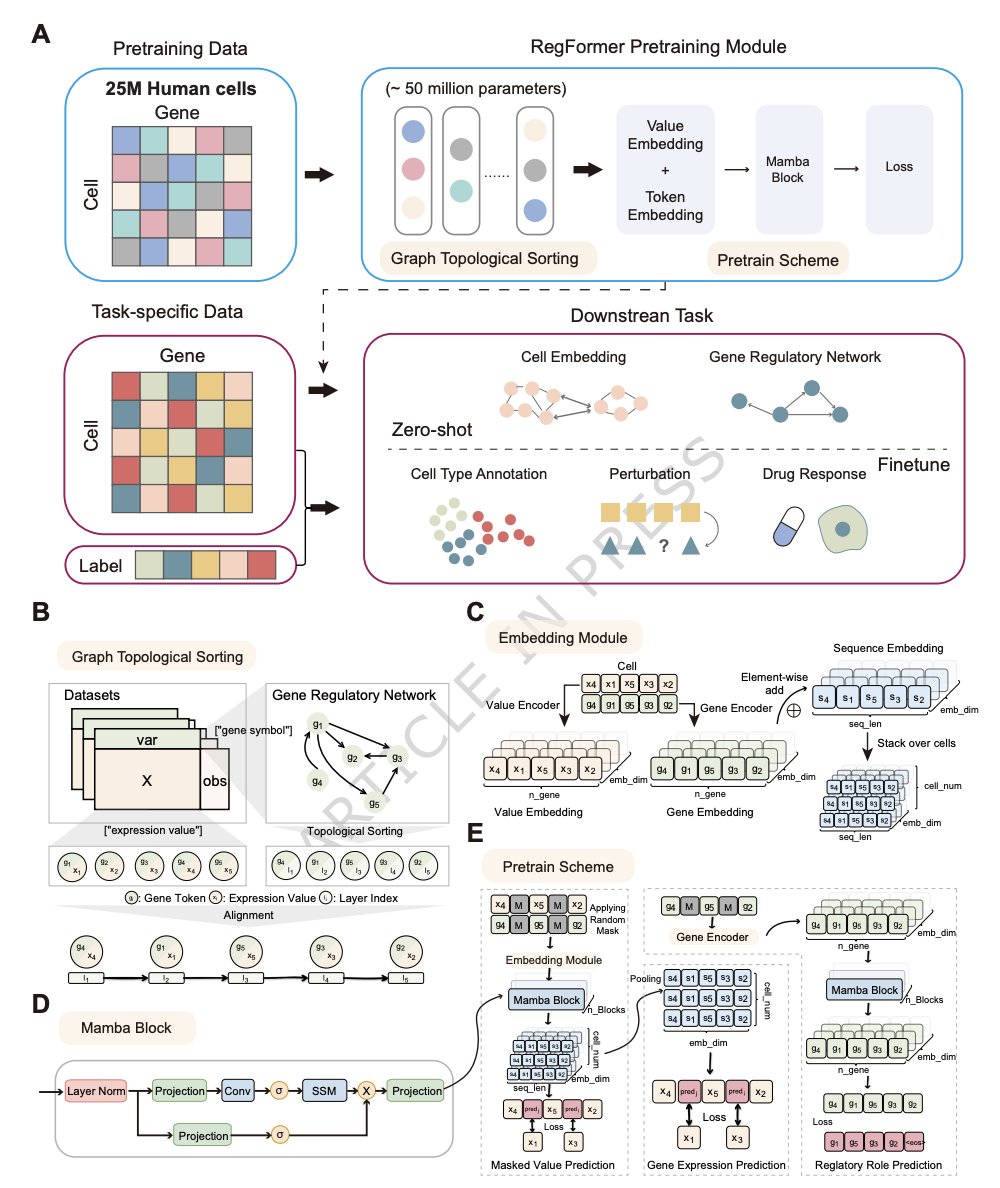
1
10
1,892
RegFormer: A single-cell foundation model powered by gene regulatory hierarchies @NatureComms
1. RegFormer is a single-cell foundation model that bakes gene regulatory hierarchies directly into the input representation: genes are ordered by a GRN-derived directed acyclic graph (DAG), so upstream regulators precede downstream targets during sequence modeling.
2. The architecture replaces Transformer attention with Mamba state-space blocks (linear-time in sequence length), enabling long-context modeling up to 10,000 genes and addressing the scalability/context-length limits common in attention-based single-cell foundation models.
3. Each gene is encoded with dual embeddings: a token embedding for gene/regulatory identity plus a value embedding for continuous expression magnitude; ablations show removing token embeddings or binarizing expression prediction degrades clustering quality, supporting the need for both regulatory context and quantitative modeling.
4. Pretraining combines three self-supervised objectives with fixed weights: masked value prediction (MVP), gene expression prediction for cells (GEPC), and regulatory topology/role prediction (TOPO). Objective ablations show the largest drop when TOPO is removed, highlighting the contribution of explicitly learning regulatory directionality.
5. Training data scale: 25 million human scRNA-seq profiles across 45 tissues and diverse technologies/conditions, harmonized via a unified pipeline and stored in LMDB for efficient streaming during large-scale pretraining.
6. GRN construction: TF–target links come from CisTarget motif-to-gene rankings (top-20 targets per TF), refined with Node2Vec embeddings and iterative edge pruning to remove cycles while retaining >92% of edges and preserving key graph statistics; genes are then topologically sorted for model input.
7. Benchmarks (BioLLM framework): across multiple tissue datasets, RegFormer yields higher Average Silhouette Width (ASW) for cell-type separation and improved batch integration compared with scGPT, Geneformer, scFoundation, and scBERT; the 10k-gene variant generally improves resolution by providing broader regulatory context.
8. Cell type annotation: RegFormer achieves higher Macro-F1 across diverse datasets, shows strong performance on fine-grained immune labels (e.g., Zheng68k), and improves rare cell type recognition; even a frozen backbone transfers well, with finetuning providing additional gains.
9. Mechanistic/biological fidelity: gene embeddings better align with GRN topology (e.g., TF vs non-TF classification; correlations with regulatory degree). GRNs reconstructed from embedding cosine similarity show higher functional coherence (GO enrichment and functional similarity), with case studies recovering canonical regulons (e.g., GATA3 in T cells; SPI1/PU.1 in myeloid cells).
10. Generalization to interventions and pharmacology: when plugged into GEARS, RegFormer embeddings support accurate CRISPR perturbation response prediction (high agreement between predicted and observed Δ-expression; responsive genes lie closer in embedding space to perturbed TFs). Integrated into DeepCDR-style pipelines, RegFormer improves IC50 prediction (PCC/SRCC) including leave-drug-out tests, and enriches pathway-level interpretability (e.g., NF-κB with Bortezomib; p53-related signals with SN-38).
💻Code: github.com/BGIResearch/RegFo…
📜Paper: doi.org/10.1038/s41467-026-7…
#scRNAseq #SingleCell #FoundationModels #GeneRegulatoryNetworks #Mamba #StateSpaceModels #Bioinformatics #ComputationalBiology #DrugResponse #CRISPR
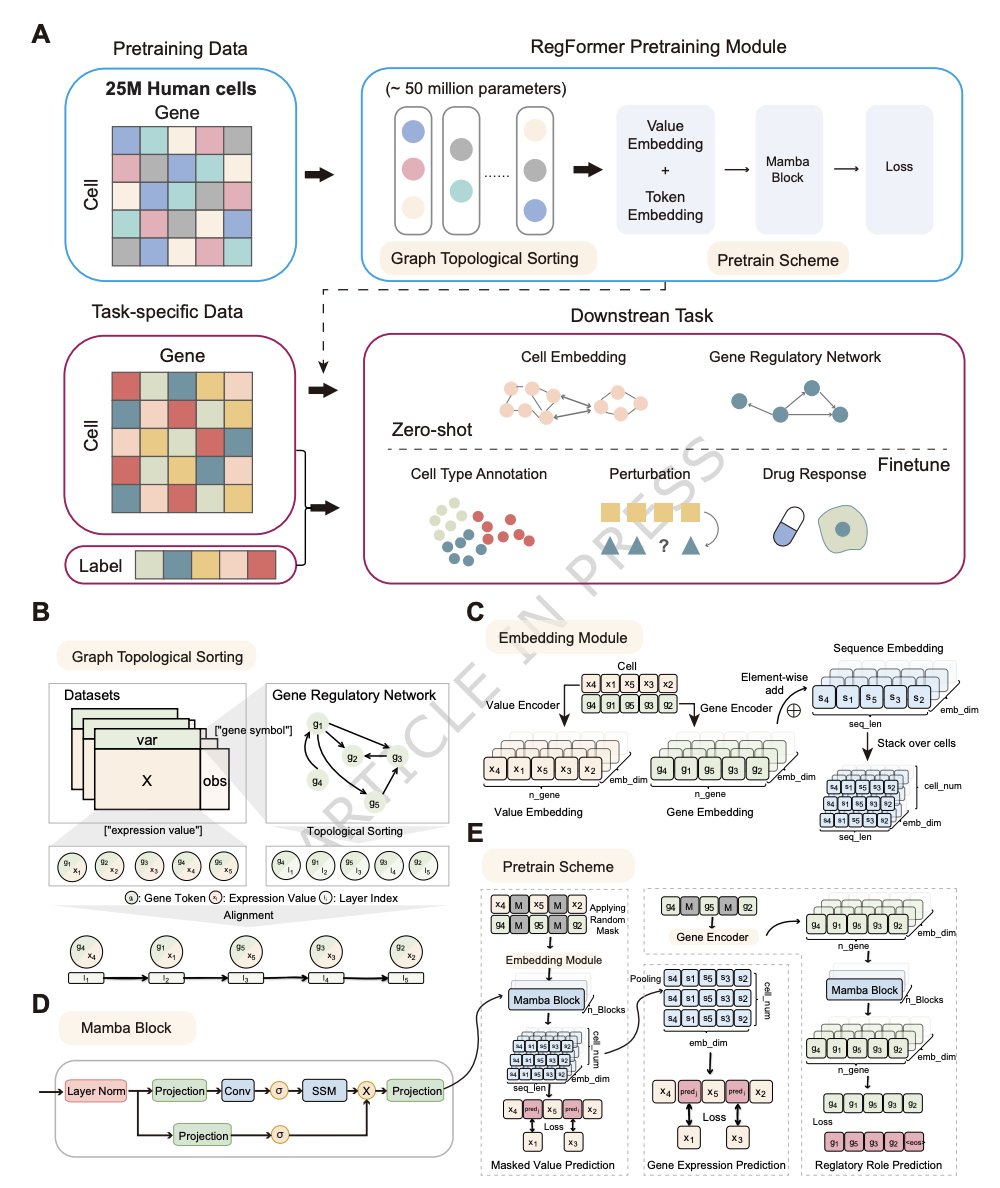
6
16
1,912

Mar 6
The biggest disruption in AI infrastructure may come from Math, not hardware.
Most AI infrastructure today is built around transformers, where compute scales O(n²) with context length.
State Space Models (SSMs) change that to O(n).
Example: context 4K → 32K tokens (8×)
• Transformer compute → 64×
• SSM compute → 8×
Infrastructure math:
Transformer GPU cost ≈ 100 units
SSM GPU cost ≈ 10–20 units
That’s roughly ~8–10× infra efficiency.
Which means:
• far fewer GPUs
• lower inference cost
• viable edge AI
Likely architecture shift:
2024–2026: Transformers MoE
2026–2028: Hybrid (Transformer SSM)
2028 : SSM-dominant models
AI advantage may move from “who has the most GPUs”
to “who designs the most efficient architectures.”
A shift from brute force → elegant design.
#AI #AIInfrastructure #LLM #StateSpaceModels #MachineLearning
1
5
113
CrossLLM-Mamba: Multimodal State Space Fusion of LLMs for RNA Interaction Prediction
1 The paper introduces CrossLLM-Mamba, a novel framework that achieves state-of-the-art performance on RNA interaction prediction by reformulating the problem as a state-space alignment task rather than static feature fusion.
2 The core innovation lies in using bidirectional Mamba encoders to enable dynamic "crosstalk" between modality-specific embeddings from biological large language models, allowing hidden states of one molecule to flow into and modulate the representation of its binding partner.
3 The framework maintains linear computational complexity, making it scalable to high-dimensional embeddings from state-of-the-art BioLLMs like ESM-2 for proteins, RiNALMo for RNA, and MoleBERT for small molecules, avoiding the quadratic scaling issues of transformer-based cross-attention.
4 On the RPI1460 benchmark for RNA-protein interactions, the model achieves an MCC of 0.892 and recall of 0.971, surpassing the previous best by 5.2% and demonstrating exceptional ability to identify true positive interactions.
5 For RNA-small molecule binding affinity prediction, CrossLLM-Mamba attains Pearson correlations exceeding 0.95 on riboswitch and repeat RNA subtypes, with consistent improvements across most RNA categories compared to existing methods.
6 The architecture incorporates Gaussian noise injection during training to enhance robustness against hard-negative samples, combined with Focal Loss to address severe class imbalance inherent in biological interaction datasets.
7 Cross-species transfer experiments on plant miRNA-lncRNA interactions show strong generalization capability, with a 7% accuracy improvement over baselines in the challenging MTR-ATH scenario, suggesting the model captures universal structural motifs conserved across species.
8 Ablation studies confirm that replacing the Cross-Mamba interaction module with simple concatenation causes the most significant performance drop, validating that modeling interaction as a dynamic state transition is superior to static feature aggregation.
📜Paper: arxiv.org/abs/2602.22236
#RNABiology #Bioinformatics #MachineLearning #StateSpaceModels #Mamba #ComputationalBiology #DrugDiscovery #MultiModalLearning #BioLLM
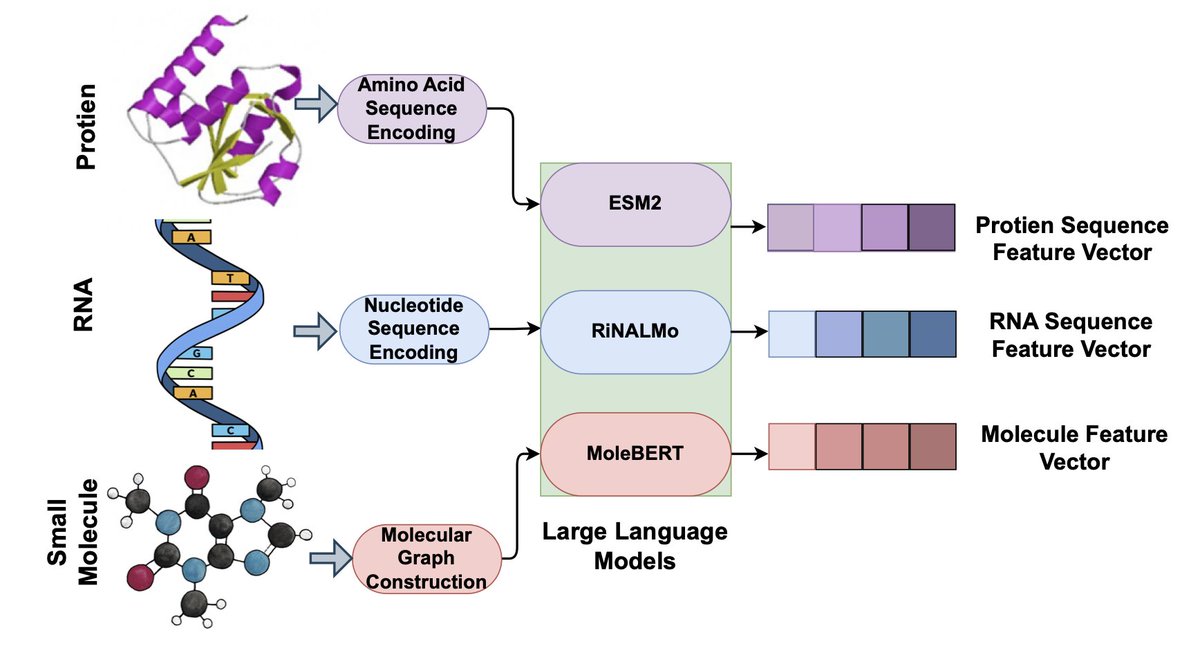
4
13
1,192

Modular 26.1 is a game-changer. We’re proud and excited to see @QWERKYAI's work featured and to be part of this innovative community! 🔥We're bringing Mamba architecture and state-space models to MAX—exploring alternatives to traditional Transformers with faster inference and much longer context windows. Our team developed custom Mojo kernels, new model architectures, and serving pipeline enhancements.
Check out more about @Modular 26.1 here:
modular.com/blog/26-1-releas…
#AI #MachineLearning #mamba #StateSpaceModels #Mojo
2
3
114

Jan 13
Full write-up in the blog post.
twosmallfish.vc/portfolio-hi…
#EdgeAI #OnDeviceAI #VoiceAI #TimeSeriesAI #Semiconductors #StateSpaceModels #Robotics #Wearables #AR #HealthTech #Automotive #VentureCapital
1
3
106

26 Aug 2025
We are delighted to share that Dr. Vandana Bharti, Assistant Professor, CSE, presented and published the paper "When Explainability Meets Vision AI: Analyzing CNNs, Transformers, and State-Space Models in Healthcare" at the 38th International Joint Conference on Neural Networks (IJCNN 2025) in Rome, Italy. Recognized as a premier global conference on AI and neural networks, IJCNN serves as a prestigious platform uniting academia and industry to showcase cutting-edge research. This achievement highlights her research excellence and international recognition in the AI community.
#IITDharwad #NeuralNetworks #IJCNN2025 #HealthcareAI #ComputerVision #Transformers #CNN #StateSpaceModels #ExplainableAI #ResearchExcellence #GlobalRecognition #MachineLearning #ArtificialIntelligence #AcademicResearch #Rome2025


3
4
15
1,059

4 Aug 2025
I wrote a blog on how to set up Mamab SSM on Kaggle
#MambaSSM #Kaggle #StructuredStateSpace #MachineLearning #DeepLearning #AIResearch #GPUComputing #StateSpaceModels #OpenSourceAI #TransformerAlternatives
ranaadeeltahir.me/blog/run-m…
1
2
109

Tired of #AI search hallucinations?
The root cause often lies in the architecture behind most AI models: the #Transformer.
This #InfoQ article explains how #StateSpaceModels (#SSMs) can fix this, & what it could mean for the future of AI search: bit.ly/4nssnWE
#LLMs

1
3
1,369

12 Jul 2025
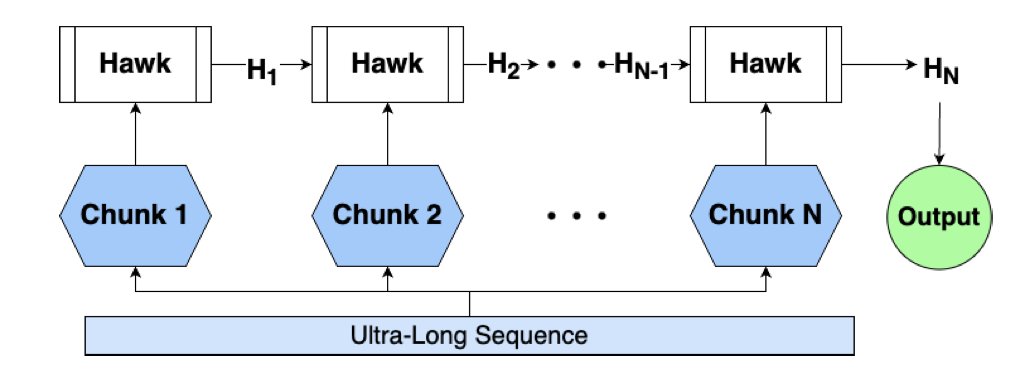
Leveraging State Space Models in Long Range Genomics
New research highlights State-Space Models (SSMs) like Caduceus and Hawk as powerful tools for genomics, overcoming the limitations of traditional transformer-based models in handling long-range dependencies. These SSMs match transformer performance and show impressive zero-shot extrapolation, processing sequences 10-100x longer than those seen during training without additional fine-tuning.
A key innovation is the ability of SSMs to efficiently process sequences of 1 million tokens on a single GPU. This allows for modeling entire genomic regions at once, which is crucial for understanding complex human genome structures and functions, even for labs with limited computational resources.
The study benchmarks Caduceus and Hawk against a 50M-parameter transformer baseline (NTv2) on various long-range genomic tasks from the GLRB benchmark. Caduceus, in particular, demonstrates competitive results, outperforming NTv2 on several tasks, showcasing the strength of SSMs in capturing long-range genomic dependencies.
SSMs exhibit superior zero-shot extrapolation capabilities compared to attention-based models. While transformers struggle with sequences longer than their training data, SSMs maintain performance with minimal degradation, indicating more generalizable representations. This is vital for genomics, where regulatory interactions can span hundreds of kilobases.
The research also shows that SSMs implicitly capture positional information through continuous hidden-state updates, providing inherent scalability and robust positional encoding. This contrasts with transformers' explicit positional encodings, which face limitations when extrapolating to extremely long genomic sequences.
The consistent masked language modeling (MLM) loss across various sequence lengths during pretraining further supports that SSMs learn high-quality internal representations capable of handling extended contexts effectively. This suggests they are learning scalable representations rather than memorizing local patterns.
📜Paper: openreview.net/forum?id=C2lf…
#Genomics #StateSpaceModels #MachineLearning #Bioinformatics #LongRangeGenomics #AI
2
639
12 Jul 2025
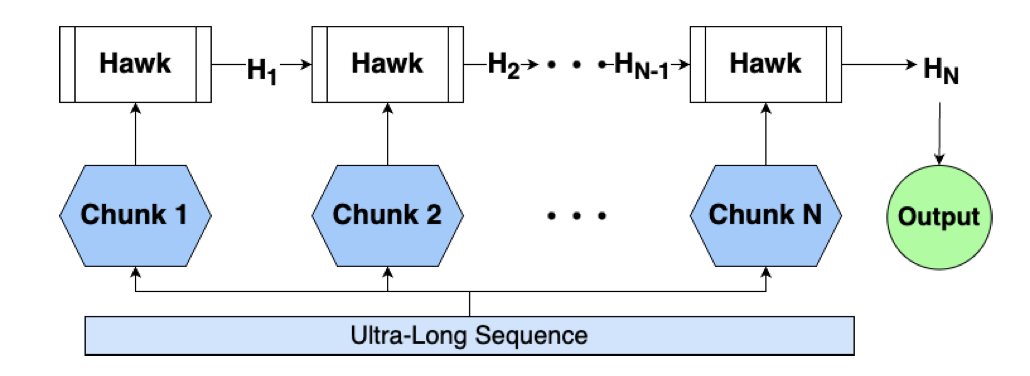
Leveraging State Space Models in Long Range Genomics
New research highlights State-Space Models (SSMs) like Caduceus and Hawk as powerful tools for genomics, overcoming the limitations of traditional transformer-based models in handling long-range dependencies. These SSMs match transformer performance and show impressive zero-shot extrapolation, processing sequences 10-100x longer than those seen during training without additional fine-tuning.
A key innovation is the ability of SSMs to efficiently process sequences of 1 million tokens on a single GPU. This allows for modeling entire genomic regions at once, which is crucial for understanding complex human genome structures and functions, even for labs with limited computational resources.
The study benchmarks Caduceus and Hawk against a 50M-parameter transformer baseline (NTv2) on various long-range genomic tasks from the GLRB benchmark. Caduceus, in particular, demonstrates competitive results, outperforming NTv2 on several tasks, showcasing the strength of SSMs in capturing long-range genomic dependencies.
SSMs exhibit superior zero-shot extrapolation capabilities compared to attention-based models. While transformers struggle with sequences longer than their training data, SSMs maintain performance with minimal degradation, indicating more generalizable representations. This is vital for genomics, where regulatory interactions can span hundreds of kilobases.
The research also shows that SSMs implicitly capture positional information through continuous hidden-state updates, providing inherent scalability and robust positional encoding. This contrasts with transformers' explicit positional encodings, which face limitations when extrapolating to extremely long genomic sequences.
The consistent masked language modeling (MLM) loss across various sequence lengths during pretraining further supports that SSMs learn high-quality internal representations capable of handling extended contexts effectively. This suggests they are learning scalable representations rather than memorizing local patterns.
@AymenKallala_ @AymenKallala_ @alainsamjr
📜Paper: openreview.net/forum?id=C2lf…
#Genomics #StateSpaceModels #MachineLearning #Bioinformatics #LongRangeGenomics #AI
3
655
30 Jun 2025
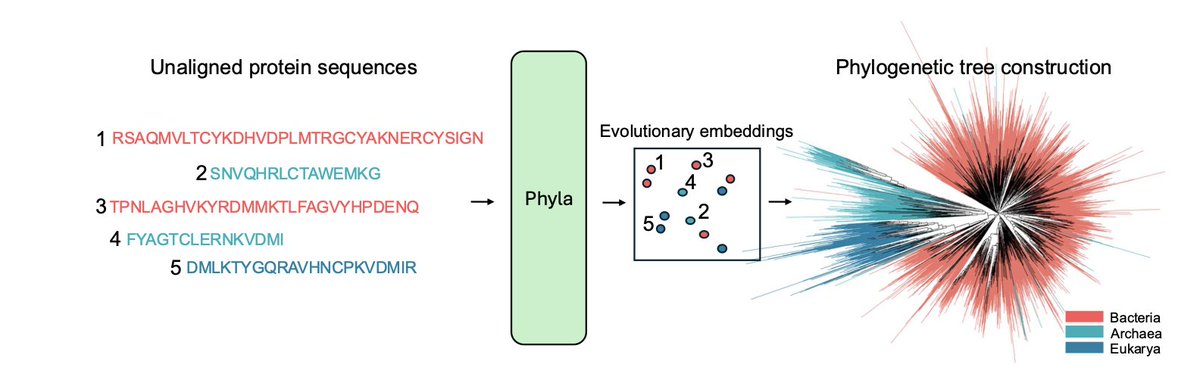
Sequence Modeling Is Not Evolutionary Reasoning
1.This paper makes a bold claim: despite widespread belief, protein language models (PLMs) do not inherently learn evolutionary relationships. Their success on tasks like masked prediction does not imply phylogenetic reasoning ability.
2.To test this, the authors introduce the first benchmark explicitly focused on evolutionary reasoning: reconstructing phylogenetic trees and clustering sequences by taxonomic lineage.
3.Surprisingly, popular PLMs such as ESM2, ESM3, and ProGen2 fail on these tasks—sometimes performing worse than a simple Hamming distance baseline. Their embeddings do not reflect true evolutionary distances.
4.To address this gap, the authors propose PHYLA: a 24M parameter hybrid state-space and transformer model that processes multiple sequences jointly and is trained with a novel tree-based loss instead of masked language modeling.
5.PHYLA is explicitly designed for inter-sequence reasoning. It alternates between modules that identify conserved motifs across sequences and ones that contextualize those motifs within each sequence.
6.The model uses quartet-based supervision, minimizing a loss that teaches the model to preserve correct evolutionary topologies in its learned embedding space—enabling alignment-free, guide-tree-free phylogenetic inference.
7.On TreeBase and TreeFam, PHYLA outperforms all tested PLMs and genomic foundation models in reconstructing trees (13.4% improvement in normRF over the next best). It also achieves the best clustering at every taxonomic level.
8.PHYLA generalizes beyond synthetic benchmarks. It reconstructs biologically coherent trees for real ribosomal proteins, correctly distinguishing functionally divergent lineages like Lokiarchaeota, and recovers known adaptations.
9.On ProteinGym, a functional prediction benchmark, PHYLA achieves a competitive Spearman correlation of 0.638—on par with much larger PLMs—showing its embeddings encode biologically meaningful signals beyond phylogeny.
10.Ablation studies reveal that both the tree-based objective and the architectural innovations (esp. sparsified attention) are essential. MLM pretraining alone is insufficient to teach evolutionary reasoning.
11.The authors argue evolutionary reasoning is not an emergent property of scaling or self-supervision on sequences. It must be directly supervised with phylogenetic signals and architectural bias—exactly what PHYLA introduces.
12.This work draws a sharp line between modeling local residue distributions (what PLMs do) and understanding evolutionary relationships (what biology needs). It points toward a new generation of biologically grounded models.
💻Code: github.com/mims-harvard/Phyl…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLanguageModels #Phylogenetics #DeepLearning #EvolutionaryBiology #Bioinformatics #MachineLearning #PLMs #Transformers #StateSpaceModels #FunctionalGenomics
4
23
2,197
30 Jun 2025
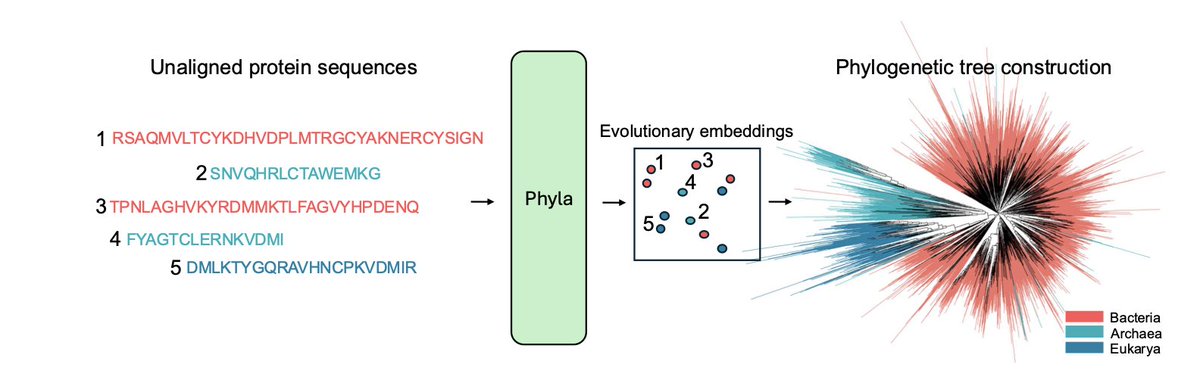
Sequence Modeling Is Not Evolutionary Reasoning
1.This paper makes a bold claim: despite widespread belief, protein language models (PLMs) do not inherently learn evolutionary relationships. Their success on tasks like masked prediction does not imply phylogenetic reasoning ability.
2.To test this, the authors introduce the first benchmark explicitly focused on evolutionary reasoning: reconstructing phylogenetic trees and clustering sequences by taxonomic lineage.
3.Surprisingly, popular PLMs such as ESM2, ESM3, and ProGen2 fail on these tasks—sometimes performing worse than a simple Hamming distance baseline. Their embeddings do not reflect true evolutionary distances.
4.To address this gap, the authors propose PHYLA: a 24M parameter hybrid state-space and transformer model that processes multiple sequences jointly and is trained with a novel tree-based loss instead of masked language modeling.
5.PHYLA is explicitly designed for inter-sequence reasoning. It alternates between modules that identify conserved motifs across sequences and ones that contextualize those motifs within each sequence.
6.The model uses quartet-based supervision, minimizing a loss that teaches the model to preserve correct evolutionary topologies in its learned embedding space—enabling alignment-free, guide-tree-free phylogenetic inference.
7.On TreeBase and TreeFam, PHYLA outperforms all tested PLMs and genomic foundation models in reconstructing trees (13.4% improvement in normRF over the next best). It also achieves the best clustering at every taxonomic level.
8.PHYLA generalizes beyond synthetic benchmarks. It reconstructs biologically coherent trees for real ribosomal proteins, correctly distinguishing functionally divergent lineages like Lokiarchaeota, and recovers known adaptations.
9.On ProteinGym, a functional prediction benchmark, PHYLA achieves a competitive Spearman correlation of 0.638—on par with much larger PLMs—showing its embeddings encode biologically meaningful signals beyond phylogeny.
10.Ablation studies reveal that both the tree-based objective and the architectural innovations (esp. sparsified attention) are essential. MLM pretraining alone is insufficient to teach evolutionary reasoning.
11.The authors argue evolutionary reasoning is not an emergent property of scaling or self-supervision on sequences. It must be directly supervised with phylogenetic signals and architectural bias—exactly what PHYLA introduces.
12.This work draws a sharp line between modeling local residue distributions (what PLMs do) and understanding evolutionary relationships (what biology needs). It points toward a new generation of biologically grounded models.
💻Code: github.com/mims-harvard/Phyl…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLanguageModels #Phylogenetics #DeepLearning #EvolutionaryBiology #Bioinformatics #MachineLearning #PLMs #Transformers #StateSpaceModels #FunctionalGenomics
1
10
1,193

9 Jun 2025
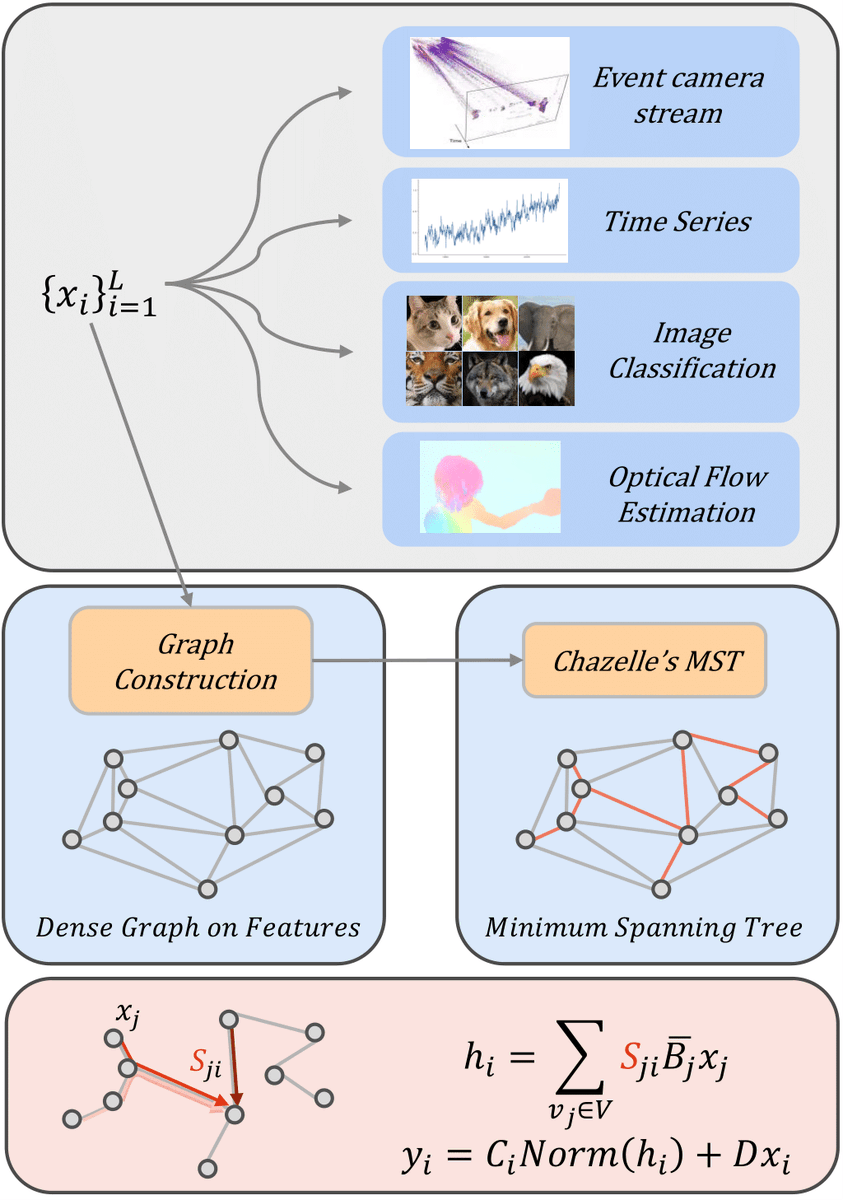
Excited to share our #CVPR2025 paper, "GG-SSMs: Graph-Generating State Space Models", to be presented as a Highlight Paper in Nashville this week!
PDF: arxiv.org/abs/2412.12423
Code: github.com/uzh-rpg/gg_ssms
While #StateSpaceModels (#SSMs) are extremely powerful for sequential data, the one-dimensional processing paradigm severely limits their ability to model non-local interactions in high-dimensional data. Even advanced models like #Mamba, #Vim, and #VMamba, though offering selective scanning, remain constrained by predetermined paths, often failing to efficiently capture the high-dimensional interactions of data dependencies.
Our new Graph-Generating State Space Models (GG-SSMs) directly address this limitation. We introduce a novel framework that dynamically constructs graphs based on inherent feature relationships, adapting to the unique structure of the data itself! By leveraging Chazelle's Minimum Spanning Tree (MST) algorithm, known for its near-linear time complexity, GG-SSMs enable robust feature propagation and efficiently model complex, long-range dependencies without prohibitive computational costs.
Our contributions are as follows:
* Integration of dynamic graph structures directly into the SSM framework, capturing complex spatial and temporal relationships.
* SOTA results across 11 diverse datasets, including ImageNet, optical flow, event-based eye tracking, and time series!
- ImageNet: Sets a new benchmark with 84.9% top-1 accuracy, outperforming prior SSMs by 1%.
- KITTI-15 Optical Flow: Achieves the lowest error rate ever reported at 2.77%.
- Eye Tracking: Improves detection rates by up to 0.33% with fewer parameters on event-based datasets.
- Time Series: Demonstrates superior forecasting accuracy across six real-world datasets.
* MST-based construction ensures linear computational complexity, making GG-SSMs highly scalable and efficient for large-scale applications.
Kudos to @NikolaZubic5 !
Reference:
Nikola Zubić and Davide Scaramuzza,
GG-SSMs: Graph-Generating State Space Models,
IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Nashville, 2025.,
PDF: arxiv.org/abs/2412.12423
Code: github.com/uzh-rpg/gg_ssms
Highlight Presentation.
@ERC_Research @uzh_ifi @UZH_en @UZH_Science
#GraphNeuralNetworks #ComputerVision #MachineLearning #AIResearch #DeepLearning #OpticalFlow #TimeSeries
3
27
2,314
5 Jun 2025
Meet us next week at #CVPR2025 in Nashville! We will present several papers on #EventCameras, #FoundationModels, and #StateSpaceModels at the main conference and workshops! I will also give a tutorial on event cameras and one on Robotics 101. Complete list with times, rooms, and links to PDFs, Code, and Videos at: docs.google.com/document/d/1…
With @hardik01shah and @NikolaZubic5
@UZH_en @UZHspacehub @UZH_Science @uzh_ifi @ERC_Research

1
2
21
2,009

9 Nov 2024
Huge thanks to my incredible co-authors @MucaCirone, @orvieto_antonio, @CristopherSalvi, and Terry Lyons!
#NeurIPS2024 #MachineLearning #DeepLearning #StateSpaceModels
🧵6/6
1
2
188

20 Oct 2024
#patter fits #StateSpaceModels to #AnimalTracking data using #ParticleFiltering and #Smoothing algorithms, generating refined #maps of #SpaceUse & #residency estimates & supporting downstream #analyses in #AcousticTelemetry systems. 2/6
2
2
5
147