
May 4
New Update SDK
peptoma-sdk v0.1.1 is now live on npm.
What's new:
1. Fixed feed response field: sequences → items with proper page & totalPages pagination.
2. Fixed filter param: diseaseTarget → disease, offset → page.
3, Fixed token balance fields: walletAddress → userId, tier → stakingTier, added earnedTotal & spentTotal.
4. Fixed leaderboard type: renamed Leaderboard → LeaderboardEntry, added username & totalContributions.
5. Added new exported types: StakingTier, StructurePrediction, SortOption, DiseaseBreakdownItem.
Updated docs: removed inaccurate model references, all examples reflect live API
npm install peptoma-sdk@latest
npmjs.com/package/peptoma-sd…
$PEPTM #DeSci
4
5
27
1,828
RosettaSearch: Multi-Objective Inference-Time Search for Protein Sequence Design
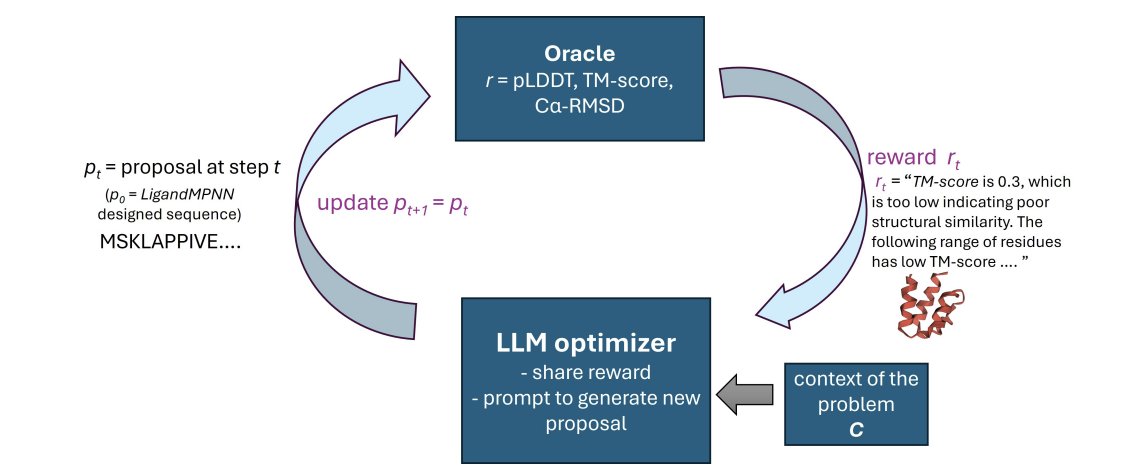
1. RosettaSearch is an inference-time framework that uses LLMs as generative optimizers to refine backbone-conditioned protein sequences via multi-objective search, guided by structural rewards from RosettaFold3, without any retraining or fine-tuning.
2. The key result: on ~400 challenging PDB monomer redesign cases initialized from suboptimal LigandMPNN outputs, RosettaSearch substantially improves structural fidelity (ss-pLDDT, TM-score, Cα-RMSD), yielding about a 2.5× increase in design success rate compared to LigandMPNN single-pass decoding.
3. The method is built around “feedback-driven editing” rather than one-shot generation: the LLM receives (a) global scalar rewards derived from structure prediction metrics and (b) local residue-range annotations highlighting problematic regions (low confidence or high deviation), then proposes targeted sequence edits.
4. Search matters: a priority-based parallel search strategy generally outperforms sequential revision. Priority search maintains a buffer of candidate sequences, repeatedly selects top-K candidates, proposes K new edits in parallel, evaluates via the structure oracle, and re-inserts all candidates—reducing premature convergence and improving robustness under a strict evaluation budget.
5. Multi-objective optimization is implemented as a weighted sum over ss-pLDDT (confidence), TM-score (global similarity), and Cα-RMSD (geometric deviation), with explicit penalties for large RMSD. Empirically, multi-objective setups improve multiple metrics simultaneously and increase success rates beyond single-objective optimization.
6. The paper emphasizes that residue-level feedback is not sufficient by itself: a compute-matched, information-matched random-mutation baseline (mutating only flagged regions) fails to improve and often degrades fidelity. The gains come from the LLM interpreting feedback and making chemically/structurally informed substitutions.
7. To reduce reward hacking (e.g., poly-alanine runs, motif repetition, length manipulation, or returning the native sequence), RosettaSearch adds soft constraints as textual feedback: repetition checks (6-mer redundancy), explicit length constraints, and (when applicable) ligand-binding position constraints to preserve functional interactions.
8. Generalization checks: improvements persist when final sequences are evaluated by an independent structure predictor (Chai-1), suggesting gains are not merely overfitting to RosettaFold3’s inductive biases. The approach also generalizes across LLM families (o4-mini and Gemini-3), and performance scales with reasoning capability (o3-mini < o4-mini).
9. Beyond native backbones, RosettaSearch also improves designs on de novo Dayhoff atlas backbones (RFDiffusion backbones with ProteinMPNN sequences). Even with strong starting success rates, RosettaSearch increases success-rate-1 from 72.4% to 89.5% and success-rate-2 from 45.5% to 67.3%, notably without providing any reference (native) sequence context.
10. A multi-modal extension uses vision-language models: rendered images of predicted structures are included as feedback to provide spatial context. Performance is comparable to text-only optimization in some settings, while producing qualitatively richer reasoning traces.
📜Paper: arxiv.org/abs/2604.17175
#ProteinDesign #ComputationalBiology #ProteinEngineering #LLM #InferenceTimeSearch #MultiObjectiveOptimization #RosettaFold #StructurePrediction #GenerativeOptimization #MachineLearning
4
6
40
2,303
RosettaSearch: Multi-Objective Inference-Time Search for Protein Sequence Design
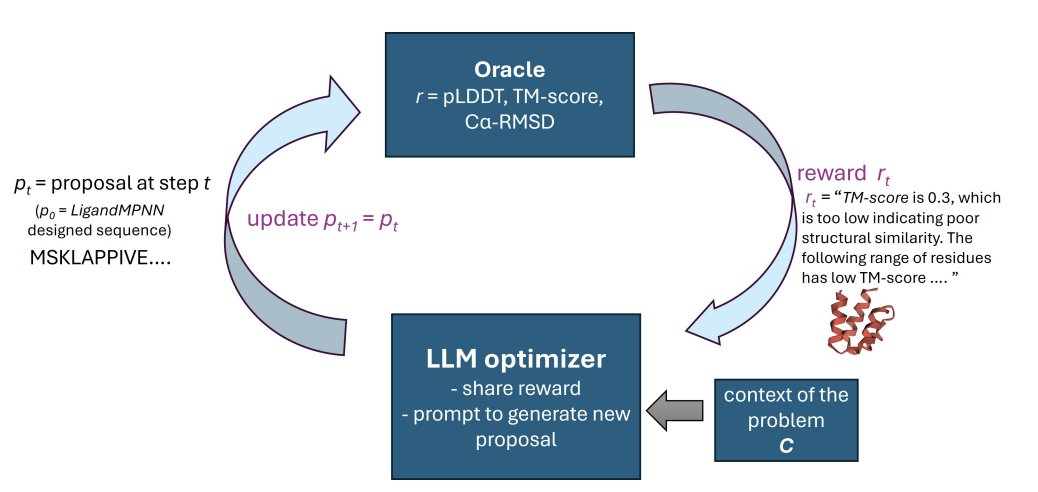
1 RosettaSearch frames backbone-conditioned protein sequence design as inference-time multi-objective optimization: an LLM proposes targeted sequence edits, while a structure predictor (RosettaFold3) provides reward detailed feedback to guide the search.
2 Key idea: don’t ask the LLM to design from scratch. Instead, start from an existing candidate (e.g., LigandMPNN output) and iteratively refine it using structured global metrics (ss-pLDDT, TM-score, Cα-RMSD) plus local residue-range annotations highlighting problematic regions.
3 The method uses a priority-based parallel search (vs. purely sequential revision): keep a buffer of evaluated candidates, repeatedly expand the top-K, propose K new variants in parallel, and reinsert all results. This reduces premature convergence and improves robustness under the same oracle-evaluation budget.
4 Multi-objective optimization is handled with a weighted-sum reward over ss-pLDDT, TM-score, and Cα-RMSD, combined with “soft constraints” delivered as text feedback to reduce reward hacking (e.g., discouraging repeats, enforcing target length, preventing trivial poly-AA solutions, and optionally preserving ligand-binding residues).
5 Large-scale evaluation on ~400 PDB monomers: starting from suboptimal LigandMPNN sequences, RosettaSearch improves structural fidelity substantially (reported improvements across metrics ranging ~18%–68%) and increases design success rate ~2.5× under RosettaFold3-based evaluation.
6 Generalization check: improvements persist when final sequences are evaluated by an independent structure oracle (Chai-1), with success rate roughly doubling vs. baselines there as well—suggesting gains aren’t purely overfitting to the feedback model.
7 Priority search vs sequential revision: both help, but priority search generally yields stronger TM-score and RMSD gains, consistent with the need to explore multiple edit trajectories in a rugged combinatorial landscape.
8 Random-mutation baseline (compute- and feedback-matched) fails to improve and often degrades designs, implying the key ingredient is not just “where to mutate” from residue-level signals, but the LLM’s ability to propose chemically/plausibly informed substitutions.
9 The approach generalizes beyond native backbones: on Dayhoff Atlas de novo backbones (ProteinMPNN initial sequences), RosettaSearch boosts success-rate-1 from 72.4% to 89.5% and success-rate-2 from 45.5% to 67.3%, without providing any reference/native sequence context.
10 Extensions and scaling: a vision-language variant can use rendered structure images as additional feedback (similar performance, richer reasoning traces). Results also generalize across LLM families (o4-mini and Gemini-3), and performance trends upward with stronger reasoning models.
📜Paper: arxiv.org/abs/2604.17175
#ProteinDesign #ComputationalBiology #LLM #ProteinEngineering #RosettaFold #StructurePrediction #Optimization #GenerativeAI #Bioinformatics
1
5
41
2,939
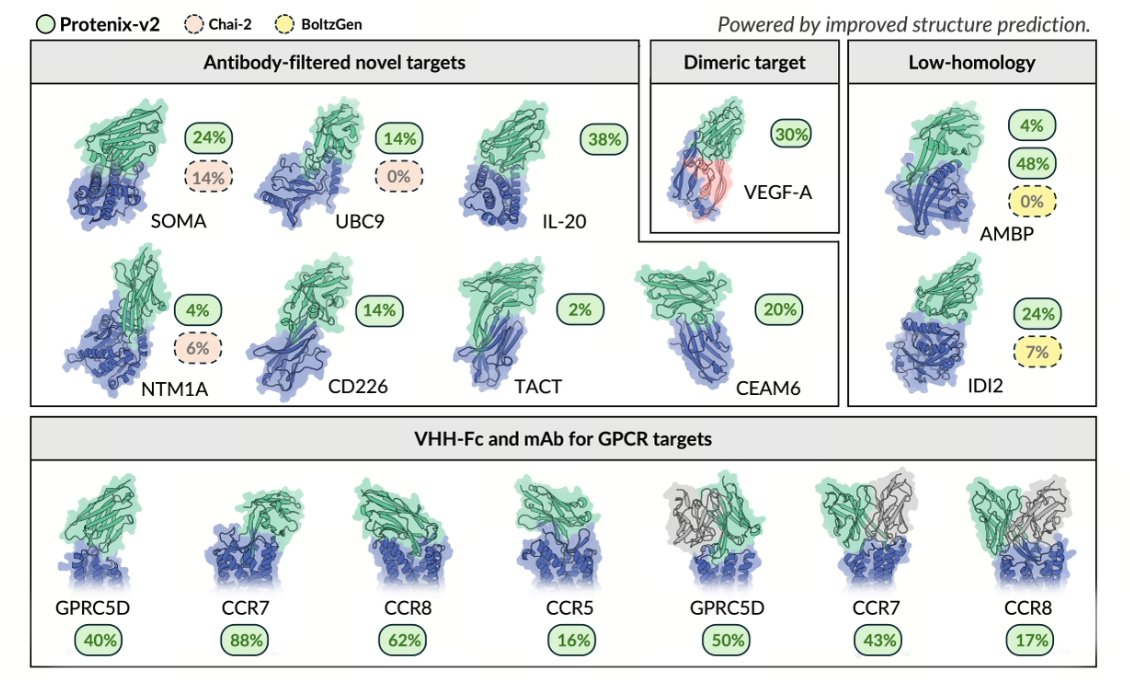
Protenix-v2: Broadening the Reach of Structure Prediction and Biomolecular Design
1. Protenix-v2 is presented as an end-to-end biomolecular modeling system spanning (a) high-accuracy structure prediction and ranking for complexes and (b) target-conditioned generative design for binders, with a focus on practical drug-discovery settings (antibodies, GPCRs, ligands, and multi-variant targets).
2. The most operationally important structure-prediction result is antibody–antigen interface modeling: across three antibody-focused benchmarks (PXMeter-AB, FoldBench-AB, AF3-AB), Protenix-v2 improves top-1 success rates at 5 seeds by 9 to 13 absolute points over Protenix-v1 at DockQ > 0.23, with similarly strong gains in the stricter DockQ > 0.8 regime.
3. A key engineering/scaling point is sampling efficiency: Protenix-v2’s 5-seed performance reportedly surpasses Protenix-v1’s 1000-seed performance, indicating much better inference-time scaling for antibody–antigen prediction (important because many pipelines rely on heavy test-time sampling).
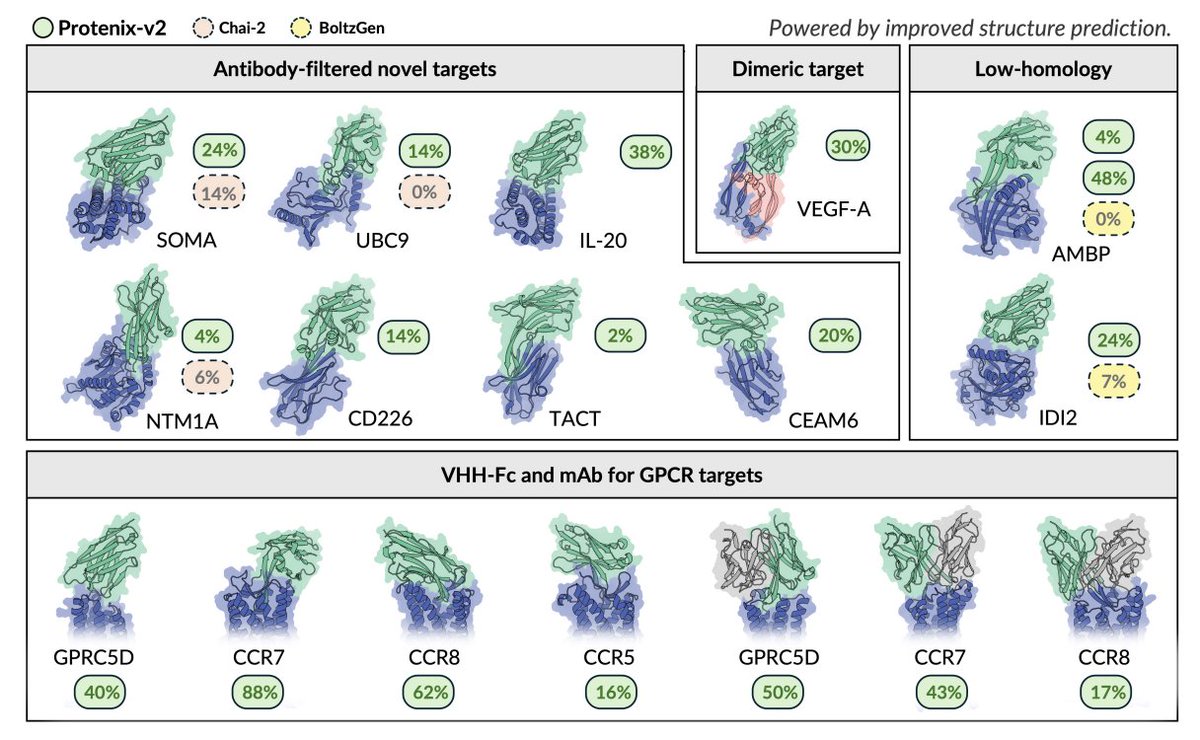
4. On zero-shot antibody design, Protenix-v2 is evaluated on a panel constructed to stress novelty and real-world constraints (in-stock antigens; novelty filtering aligned to prior work; inclusion of a dimeric case study; and multiple GPCR membrane targets). In novelty-controlled VHH-Fc campaigns, it achieves a 100% target-level success rate in the tested panel, with BLI-confirmed hit rates ranging from 2% to 48%.
5. The paper emphasizes that epitope choice can dominate difficulty even on the same antigen: for AMBP, two different epitope choices yielded hit rates of 4% vs 48%, motivating alignment of epitope selection with native ligand-binding interfaces for novelty-filtered comparisons.
6. For challenging GPCRs (small, flexible extracellular epitopes), Protenix-v2 is tested under tight experimental budgets (16–30 designs per target) and still yields BLI-confirmed hits in both VHH-Fc and full-length mAb formats: VHH-Fc hit rates of 16%–88% across four GPCR targets and mAb hit rates up to 50% (with variability across targets). For GPRC5D, the best VHH-Fc KD is reported as 112 pM (measured under avidity conditions due to antigen dimerization).
7. Developability is treated as a first-class outcome rather than an afterthought: reported pass rates on experimental assays are 100% (thermostability, DSF), 98% (self-interaction, AC-SINS), and 93% (polyreactivity, BVP ELISA). Hits also span multiple structural clusters (framework-aligned RMSD clustering), suggesting the model does not collapse onto a single binding mode.
8. The system includes model-based rankers that can select binders beyond human intuition in a VEGF-A (dimeric) case study: two integrated rankers each pick 30 candidates from ~300 designs and recover 9–10 binders (with partial overlap), while a human expert recovers 7 binders that are all unique and non-overlapping with model selections—implying complementary selection biases (models skew toward higher response; human toward broader cluster diversity).
9. Beyond antibodies, Protenix-v2 adds a ligand-geometry realism upgrade via training-free guidance (TFG): it introduces stricter “revised validity” checks (e.g., sp2 planarity, amide planarity, sp3 non-planarity) on top of PoseBusters-style criteria, and uses constraint-like guidance (chirality/planarity/torsions/distances) to improve joint success (pocket-aligned ligand RMSD < 2 Å plus validity). On PXM-22to25-Ligand, Protenix-v2-TFG reaches 60.46% under the revised criterion vs 53.96% for Boltz-1x, and is close to Boltz-2x (noted to have potential training overlap due to a later cutoff).
10. The paper also demonstrates breadth-oriented mini-binder design across SARS-CoV-2 variants: when prototype and Omicron B.1.1.529 RBDs are jointly provided, 2 of 4 tested designs bind both variants with nanomolar-scale KD (e.g., ~201–250 nM for prototype and ~146–148 nM for Omicron), with proposed compensatory interface interactions around the Q493R mutation.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #ProteinDesign #AntibodyDesign #StructurePrediction #GPCR #DrugDiscovery #DiffusionModels #ProteinLigand #Bioinformatics #MachineLearning
1
4
5
1,002
Protenix-v2: Broadening the Reach of Structure Prediction and Biomolecular Design
1. Protenix-v2 is presented as an end-to-end biomolecular modeling system spanning (a) high-accuracy structure prediction and ranking for complexes and (b) target-conditioned generative design for binders, with a focus on practical drug-discovery settings (antibodies, GPCRs, ligands, and multi-variant targets).
2. The most operationally important structure-prediction result is antibody–antigen interface modeling: across three antibody-focused benchmarks (PXMeter-AB, FoldBench-AB, AF3-AB), Protenix-v2 improves top-1 success rates at 5 seeds by 9 to 13 absolute points over Protenix-v1 at DockQ > 0.23, with similarly strong gains in the stricter DockQ > 0.8 regime.
3. A key engineering/scaling point is sampling efficiency: Protenix-v2’s 5-seed performance reportedly surpasses Protenix-v1’s 1000-seed performance, indicating much better inference-time scaling for antibody–antigen prediction (important because many pipelines rely on heavy test-time sampling).
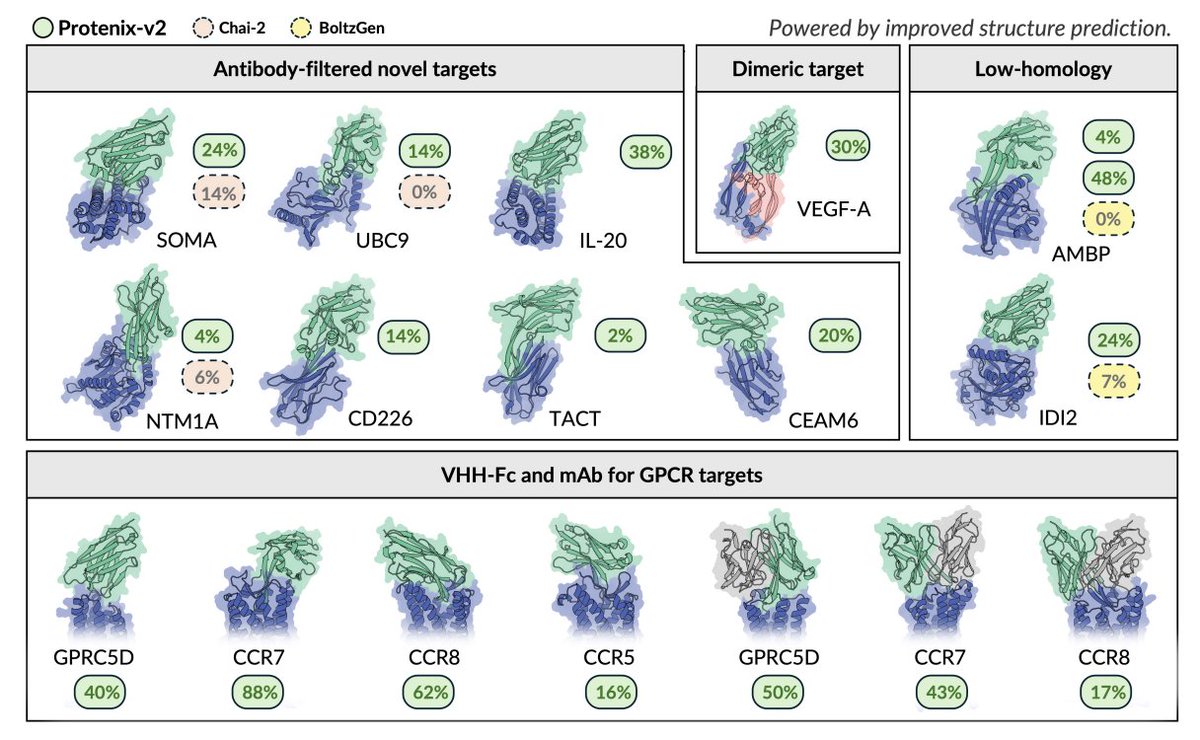
4. On zero-shot antibody design, Protenix-v2 is evaluated on a panel constructed to stress novelty and real-world constraints (in-stock antigens; novelty filtering aligned to prior work; inclusion of a dimeric case study; and multiple GPCR membrane targets). In novelty-controlled VHH-Fc campaigns, it achieves a 100% target-level success rate in the tested panel, with BLI-confirmed hit rates ranging from 2% to 48%.
5. The paper emphasizes that epitope choice can dominate difficulty even on the same antigen: for AMBP, two different epitope choices yielded hit rates of 4% vs 48%, motivating alignment of epitope selection with native ligand-binding interfaces for novelty-filtered comparisons.
6. For challenging GPCRs (small, flexible extracellular epitopes), Protenix-v2 is tested under tight experimental budgets (16–30 designs per target) and still yields BLI-confirmed hits in both VHH-Fc and full-length mAb formats: VHH-Fc hit rates of 16%–88% across four GPCR targets and mAb hit rates up to 50% (with variability across targets). For GPRC5D, the best VHH-Fc KD is reported as 112 pM (measured under avidity conditions due to antigen dimerization).
7. Developability is treated as a first-class outcome rather than an afterthought: reported pass rates on experimental assays are 100% (thermostability, DSF), 98% (self-interaction, AC-SINS), and 93% (polyreactivity, BVP ELISA). Hits also span multiple structural clusters (framework-aligned RMSD clustering), suggesting the model does not collapse onto a single binding mode.
8. The system includes model-based rankers that can select binders beyond human intuition in a VEGF-A (dimeric) case study: two integrated rankers each pick 30 candidates from ~300 designs and recover 9–10 binders (with partial overlap), while a human expert recovers 7 binders that are all unique and non-overlapping with model selections—implying complementary selection biases (models skew toward higher response; human toward broader cluster diversity).
9. Beyond antibodies, Protenix-v2 adds a ligand-geometry realism upgrade via training-free guidance (TFG): it introduces stricter “revised validity” checks (e.g., sp2 planarity, amide planarity, sp3 non-planarity) on top of PoseBusters-style criteria, and uses constraint-like guidance (chirality/planarity/torsions/distances) to improve joint success (pocket-aligned ligand RMSD < 2 Å plus validity). On PXM-22to25-Ligand, Protenix-v2-TFG reaches 60.46% under the revised criterion vs 53.96% for Boltz-1x, and is close to Boltz-2x (noted to have potential training overlap due to a later cutoff).
10. The paper also demonstrates breadth-oriented mini-binder design across SARS-CoV-2 variants: when prototype and Omicron B.1.1.529 RBDs are jointly provided, 2 of 4 tested designs bind both variants with nanomolar-scale KD (e.g., ~201–250 nM for prototype and ~146–148 nM for Omicron), with proposed compensatory interface interactions around the Q493R mutation.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #ProteinDesign #AntibodyDesign #StructurePrediction #GPCR #DrugDiscovery #DiffusionModels #ProteinLigand #Bioinformatics #MachineLearning
3
25
2,607
Protenix-v2: A Biomolecular Modeling System for Structure Prediction and Zero-Shot Antibody Design @ai4s_protenix
1. Protenix-v2 achieves massive gains in antibody-antigen structure prediction, with up to 13-point improvements over Protenix-v1 at DockQ >0.23 and comparable gains at the stricter DockQ >0.8 threshold. Most remarkably, its 5-seed performance surpasses previous 1000-seed results, representing a dramatic leap in sampling efficiency.
2. The system demonstrates 100% target-level success rate in zero-shot VHH antibody design across novelty-controlled targets, with BLI-confirmed hit rates ranging from 2% to 48%. The resulting hits show exceptional developability with 100% thermostability pass rate, 98% self-interaction pass rate, and 93% polyreactivity pass rate.
3. On challenging GPCR targets with small and flexible exposed epitopes, Protenix-v2 achieves hit rates of 16%-88% in VHH-Fc format and up to 50% in mAb format, despite testing only 16-30 designs per target. This demonstrates effective sample efficiency on difficult membrane proteins.
4. The model introduces training-free guidance (TFG) variants that significantly improve ligand-related plausibility, reaching 60.46% success rate on recent protein-ligand benchmarks under a revised stricter validity criterion that checks planarity around sp2 centers and non-planarity at sp3 centers.
5. Protenix-v2 successfully designs dual-specific binders against both prototype and Omicron SARS-CoV-2 RBD variants with nanomolar-scale KD, showing potential compensatory mechanisms at the structural level to accommodate sequence differences.
6. The system supports flexible, target-conditioned generation with granular control over CDR loop lengths and integration of predefined frameworks, spanning diverse formats from miniproteins to VHH and full-length antibodies.
💻Code: github.com/bytedance/Proteni…
📜Paper: github.com/bytedance/Proteni…
#ProtenixV2 #AntibodyDesign #StructurePrediction #AlphaFold3 #BiomolecularModeling #DrugDiscovery #GPCR #MachineLearning #ComputationalBiology #ZeroShotDesign
38
124
18,462
Enhancing anticancer peptide discovery: A fusion-centric framework with conditional diffusion for prediction and generation
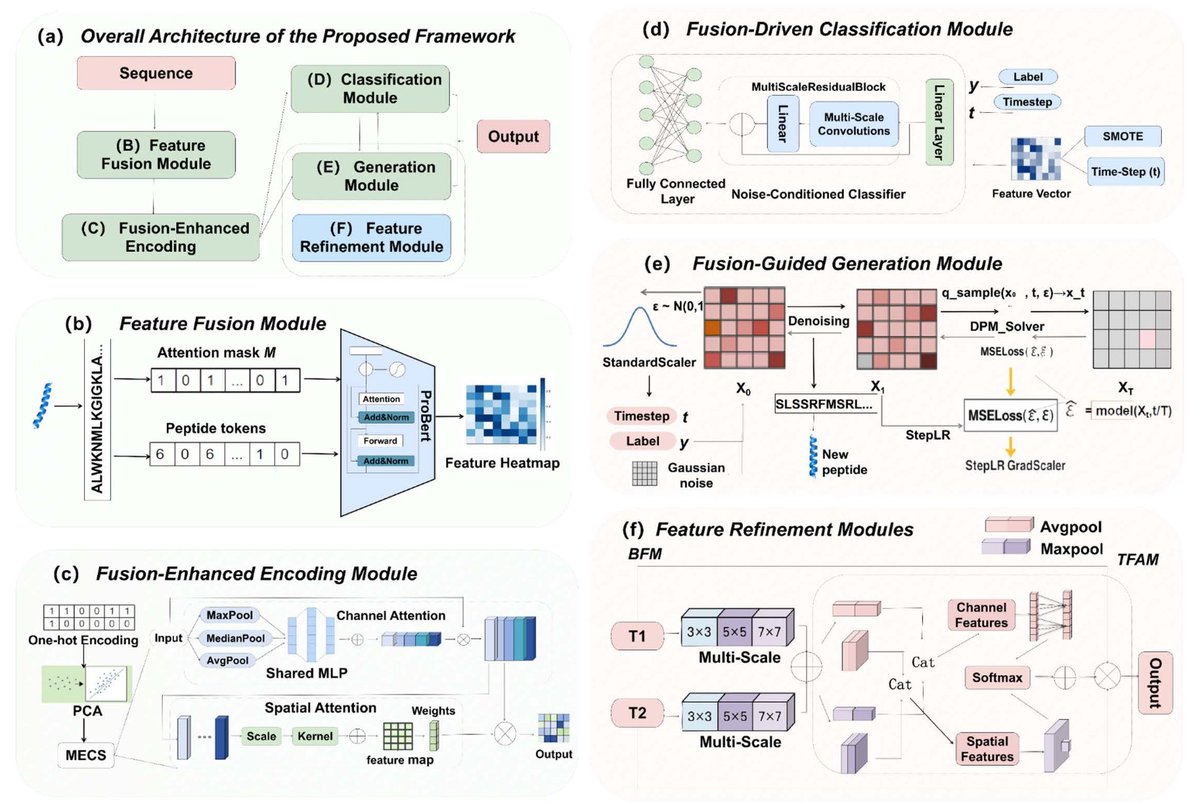
1 UACD-ACPs presents a unified pipeline that does both ACP classification and cancer-type-aware peptide generation, aiming to close a practical gap in the field where prediction models rarely connect to controllable design plus downstream structural/biophysical validation.
2 The key classification idea is “diffusion-inspired” noise conditioning: instead of running a full diffusion model for classification, the encoder injects a stochastic time-step embedding (plus cancer-type label embeddings) into fused peptide features to learn smoother, more robust decision boundaries—especially under imbalanced multi-class labels.
3 Representation is explicitly fusion-centric: ProtBERT semantic embeddings (1024-d) are concatenated with physicochemical descriptors (430-d; e.g., composition, dipeptides, pI, hydrophobicity), creating a heterogeneous feature space intended to capture both sequence semantics and interpretable biochemical signals.
4 MECS (Multiscale Embedding Compression Strategy) is introduced to reduce redundancy while preserving global-local cues: it combines channel attention (avg/max/median pooling shared MLP-like convs) with spatial attention using multiscale depthwise/asymmetric convolutions (e.g., 1×7, 7×1, 1×11, 11×1, 1×21, 21×1) to highlight functionally important regions.
5 Class imbalance is handled with a pragmatic pairing: SMOTE oversampling at the minibatch level plus class-weighted cross-entropy, then reinforced by the noise-conditioned training signal that acts like regularization against overfitting to majority classes.
6 On a 9-class cancer-type ACP dataset (e.g., breast, cervical, colon, gastric, HCC, leukemia, lung, prostate, histiocytic lymphoma), the classifier reports strong multi-class performance: AUC-ROC 0.99 ± 0.01, accuracy 0.94 ± 0.01, F1 0.93 ± 0.01, recall 0.93 ± 0.01, outperforming baselines such as ACPScanner, mACPpred, ANNprob-ACPs, XGBoost, and LightGBM.
7 The generation module uses a conditional denoising diffusion model to synthesize peptide representations that are later decoded into sequences of length 8–50, with outputs organized by cancer type to support targeted downstream screening rather than “one-bucket” generic ACP generation.
8 Two generation-specific fusion blocks are proposed to improve biological fidelity: BFM (Bitemporal Fusion Module) for multi-receptive-field feature capture across temporal states, and TFAM (Temporal Feature Attention Module) that applies channel spatial attention across two temporal branches and fuses them with softmax-weighted interactions.
9 The paper emphasizes multi-layer validation beyond sequence metrics: BLAST local alignments to check non-trivial similarity patterns, physicochemical profiling (charge, hydrophobicity, instability, aromaticity), disorder prediction (IUPred2A), structure modeling (AlphaFold2; pLDDT > 70 treated as reasonable for short peptides), and secondary-structure annotation (DSSP).
10 Biophysical plausibility is probed with simulations and docking: 100 ns all-atom MD (GROMACS, CHARMM36, DOPC:POPS membrane) for peptide–membrane interaction stability; plus qualitative HER2 kinase-domain docking (HawkDock/HDOCK) to compare feasible binding poses and residue contacts, framed as prioritization evidence rather than affinity claims.
💻Code: github.com/yidingneng/ACP-Co…
📜Paper: doi.org/10.1371/journal.pcbi…
#ComputationalBiology #Bioinformatics #PeptideDesign #AnticancerPeptides #DiffusionModels #ProteinLanguageModels #DeepLearning #DrugDiscovery #MolecularDynamics #StructurePrediction
2
12
1,322
SeekRBP: Leveraging Sequence-Structure Integration with Reinforcement Learning for Receptor-Binding Protein Identification
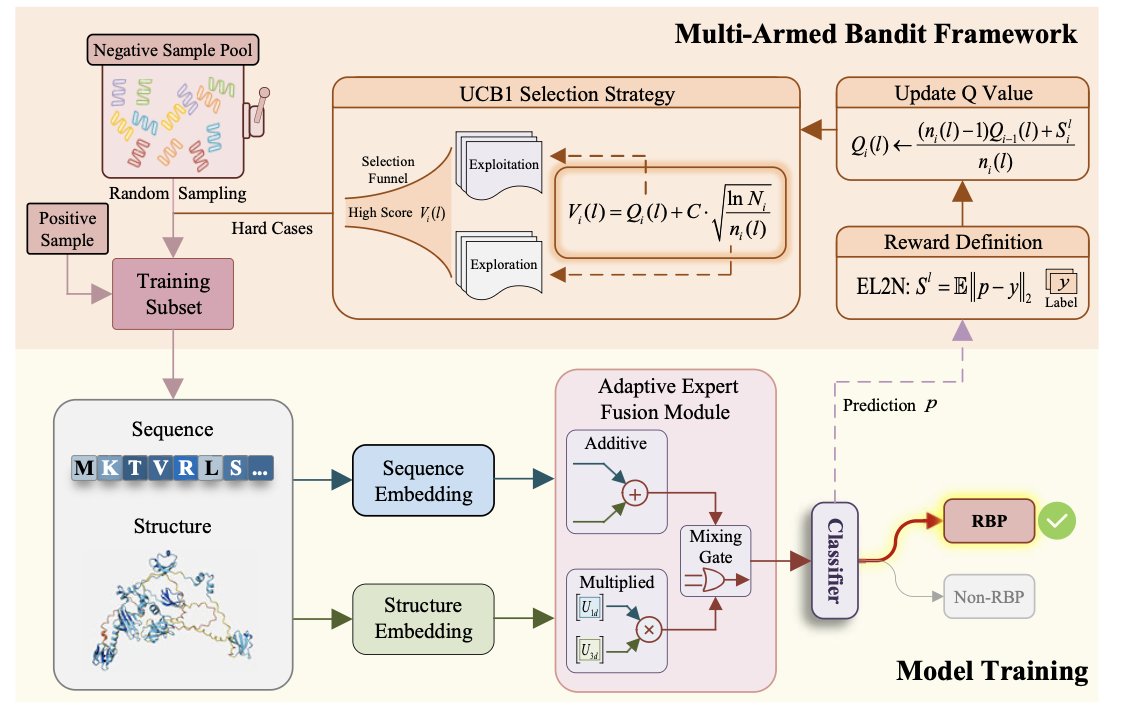
1 The paper tackles the long‑standing problem of identifying receptor‑binding proteins (RBPs) in phage genomes, where extreme sequence divergence and severe class imbalance make traditional homology searches unreliable. SeekRBP introduces a novel framework that treats RBP discovery as a sequential decision problem, allowing the model to learn from the most informative examples rather than a static training set.
2 A key innovation is the use of a multi‑armed bandit (UCB1) strategy to perform adaptive negative sampling. By continuously rewarding hard negatives that the model misclassifies, SeekRBP focuses learning on challenging non‑RBPs, avoiding over‑reliance on easy negatives and improving generalization across diverse phage families.
3 The architecture employs a dual‑branch encoder: an ESM2 protein language model extracts sequence embeddings, while ColabFold predictions followed by Saprot produce structure‑aware representations. This combination captures both the evolutionary signal in the amino‑acid sequence and the conserved three‑dimensional motifs that underpin receptor binding.
4 To fuse these heterogeneous modalities, SeekRBP introduces an Adaptive Expert Fusion Module that balances additive and low‑rank multiplicative interactions through a lightweight gating mechanism. This adaptive fusion dynamically weights each interaction type per sample, yielding richer cross‑modal representations without inflating model size.
5 Benchmark experiments on a rigorously curated dataset show SeekRBP achieving an AUROC of 0.9418, surpassing state‑of‑the‑art machine‑learning tools (PhANNs, PhageRBPdetection) and homology methods (BLASTp, Pharokka). The performance gap is especially pronounced at low false‑positive rates, demonstrating the efficacy of the bandit‑based sampling.
6 In a real‑world case study on Vibrio phages, SeekRBP identified a broader set of RBPs than manual structural annotations, and the predicted proteins improved downstream host‑prediction accuracy. Structural validation with AlphaFold3 confirmed that many of the novel RBPs adopt plausible binding conformations, underscoring the biological relevance of the predictions.
7 The framework offers a scalable, annotation‑prone solution for phage research, with immediate implications for phage therapy design and synthetic biology. Future work aims to extend the model to multi‑class receptor specificity and incorporate richer interaction dynamics for even finer host‑range predictions.
💻Code: github.com/Saillxl/SeekRBP
📜Paper: arxiv.org/abs/2603.04748
#phageomics #proteinannotation #deeplearning #bioinformatics #RBPs #reinforcementlearning #structureprediction #phagetherapy #syntheticbiology
2
9
1,038
Structural Plausibility Without Binding Specificity: Limits of AI-Based Antibody-Antigen Structure Prediction Confidence Scores
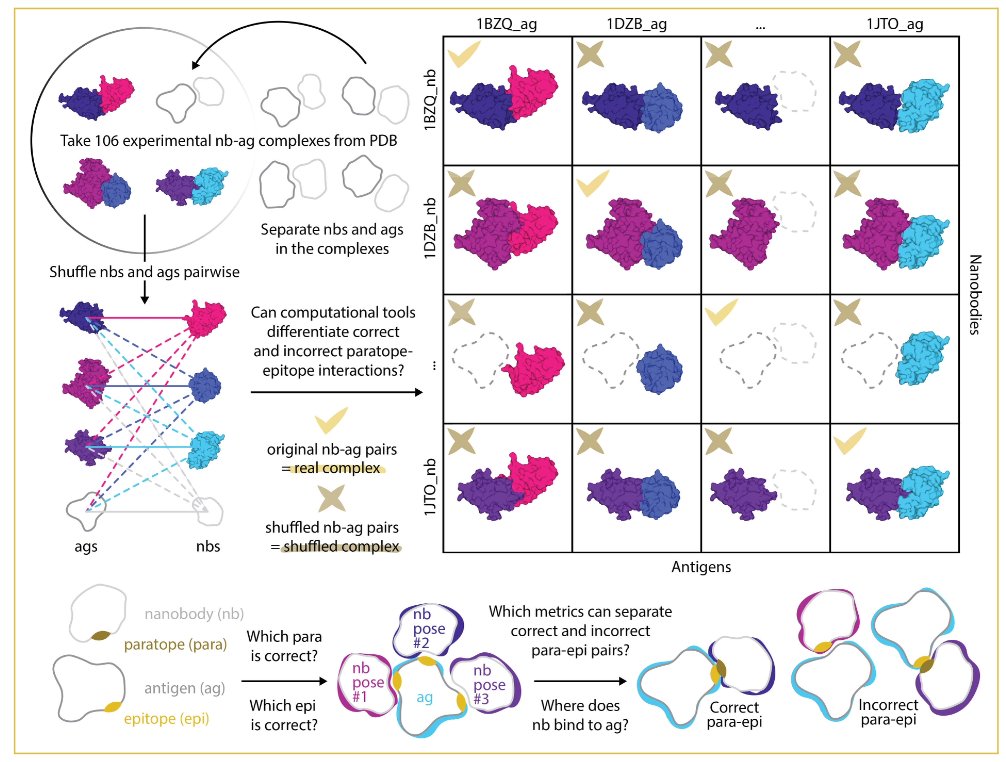
1. A sobering reality check on AI structure prediction: AlphaFold3, Boltz2, and Chai1 generate geometrically plausible antibody-antigen complexes even for biologically incorrect pairings, with confidence scores (ipTM) failing to distinguish real from shuffled interactions.
2. The study introduces a rigorous "real vs shuffled" benchmark using 106 experimentally determined nanobody-antigen complexes and 11,342 artificial non-cognate pairings, exposing how internal confidence metrics lack specificity for true biological binding.
3. Precision-recall analysis reveals poor discrimination: AF3 achieves the highest PR-AUC at merely 0.187, Boltz2 collapses to 0.026 with uniformly high false positives, and Chai1 reaches only 0.067—far from reliable screening tools.
4. Saturation sampling improves structural quality (DockQ) but critically fails to improve confidence calibration—ΔDockQ and ΔipTM show near-zero correlation across all models, indicating confidence scores remain "locked in" to early trajectory choices.
5. The research demonstrates that increased computational investment in deeper sampling yields diminishing returns for specificity, while distributing resources across independent seeds proves more informative for exploring solution landscapes.
6. Epitope recovery analysis shows models frequently identify plausible antigen contact regions even in non-cognate pairings, with shuffled complexes achieving comparable epitope recall to real complexes—geometric plausibility does not imply biological correctness.
7. Cross-tool agreement on confidence scores is remarkably weak (Pearson r = 0.13-0.18), with high-confidence outliers rarely overlapping between models, underscoring that confident failures are tool-specific rather than universal.
8. Energy efficiency analysis reveals AF3 consumes 3.6x less GPU energy than Chai1 at saturation, yet all models show optimal quality gains per watt at modest sampling depths (N=10-25), arguing against deep sampling as a default strategy.
9. The authors propose a fundamental reframing of discovery workflows: separating geometry confidence, mode confidence, and specificity confidence—moving toward comparative evaluation against realistic decoys rather than absolute thresholding.
10. Key practical guidance emerges: use shallow ensembles for cost-effective exploration, treat sampling as mode detection not confidence amplification, and never interpret ipTM as probability of correct binding without target-specific calibration against explicit negatives.
💻Code: github.com/csi-greifflab/ab_…
📜Paper: biorxiv.org/content/10.64898…
#AlphaFold3 #Boltz2 #Chai1 #AntibodyDesign #StructurePrediction #ComputationalBiology #DrugDiscovery #AIforScience #Biophysics #MachineLearning
11
43
3,634
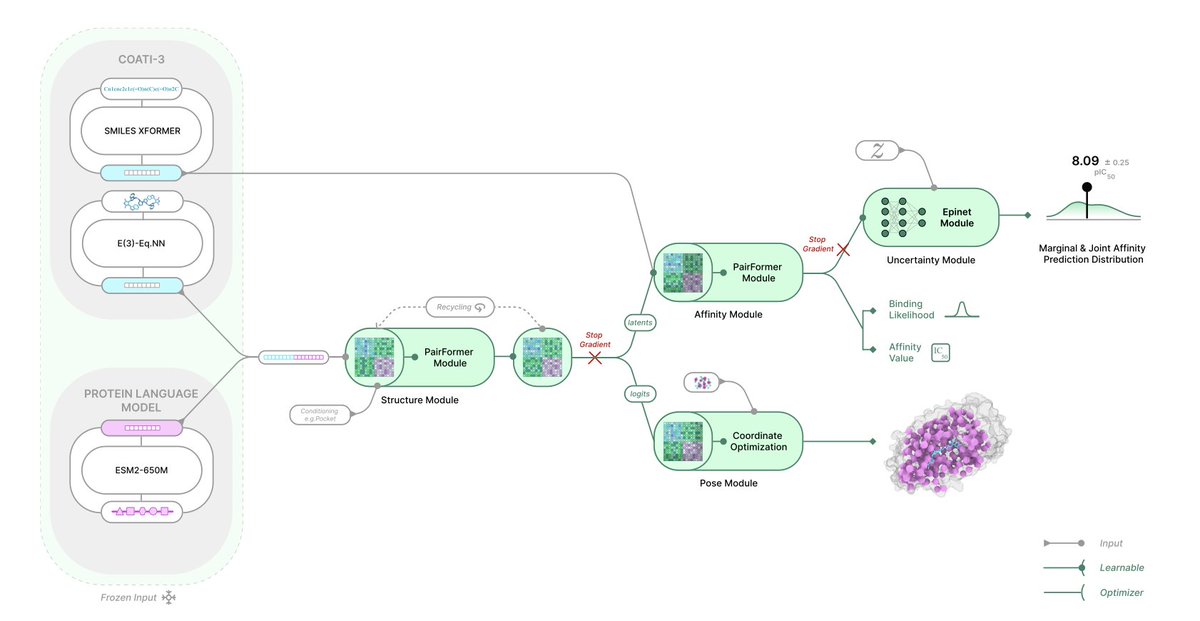
TerraBind: Fast and Accurate Binding Affinity Prediction through Coarse Structural Representations
1. TerraBind achieves 26-fold faster inference than state-of-the-art methods like Boltz-2 while improving binding affinity prediction accuracy by approximately 20%, addressing a critical computational bottleneck in structure-based drug design.
2. The core innovation challenges the prevailing assumption that full all-atom diffusion is necessary for accurate predictions. Instead, TerraBind uses a coarse pocket-level representation with only protein Cβ atoms and ligand heavy atoms, eliminating expensive generative modeling.
3. The architecture combines frozen pretrained encoders—COATI-3 for molecular representations and ESM-2 for protein sequences—with a lean 48-layer pairformer trunk of just 27M parameters, compared to Boltz-2's 509M parameters.
4. For pose generation, TerraBind employs a diffusion-free optimization module that produces 3D coordinates in under 0.2 seconds, matching diffusion-based baselines on FoldBench, PoseBusters, and Runs N'Poses benchmarks.
5. The binding affinity module operates directly on structural pairformer representations without requiring coordinate generation, outperforming Boltz-2 on 15 of 18 proprietary drug discovery targets and achieving superior Pearson correlation on CASP16.
6. A built-in uncertainty quantification system uses pairwise distance entropy as a zero-shot confidence metric, validated to correlate with both pose accuracy and binding strength without separate training.
7. The epistemic neural network (epinet) module provides calibrated affinity uncertainty estimates, enabling a continual learning framework that achieves 6× greater affinity improvement over greedy selection strategies in simulated drug discovery cycles.
8. Structural fine-tuning on minimal proprietary crystallographic data (as few as 3-6 structures) yields 17% affinity improvement on held-out compounds, demonstrating practical adaptability for specific drug programs.
📜Paper: arxiv.org/abs/2602.07735
#TerraBind #DrugDiscovery #MachineLearning #ProteinLigand #BindingAffinity #StructurePrediction #ComputationalBiology #AIforScience
1
13
47
2,853

3 Dec 2025
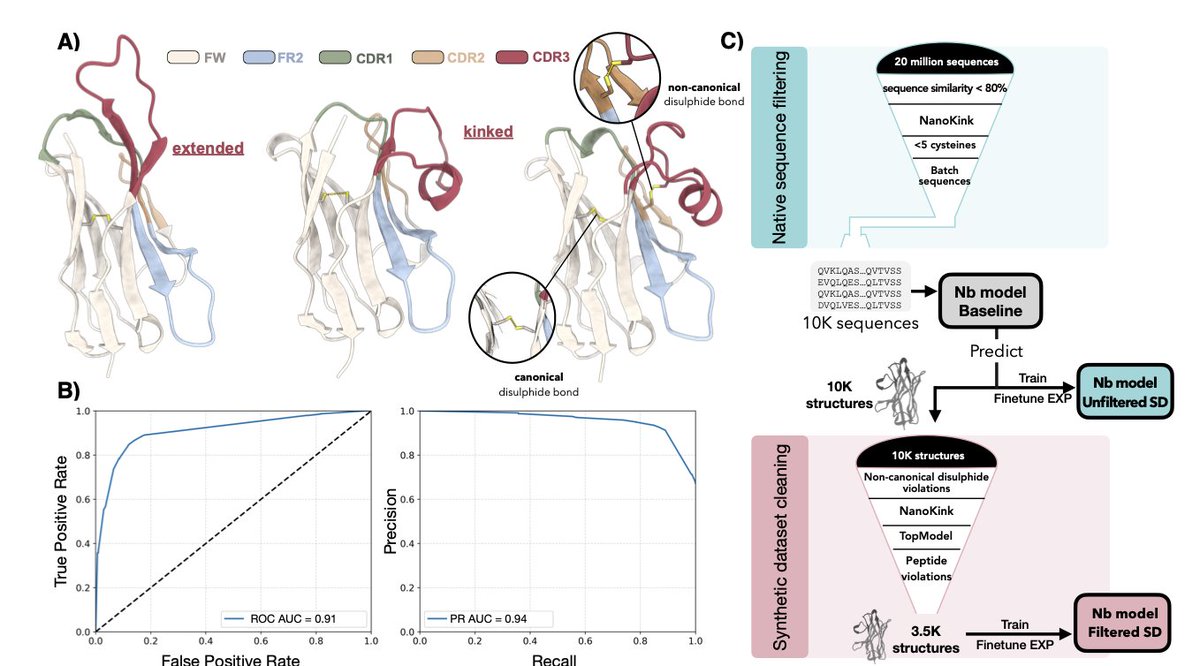
Improving nanobody structure prediction with self-distillation
1. A new study explores enhancing nanobody structure prediction using self-distillation and synthetic data curation. The authors develop a method called NanoKink to classify H-CDR3 loop conformations as kinked or extended from sequence information alone, significantly improving prediction accuracy.
2. The research highlights the challenges of predicting nanobody H-CDR3 loops due to their unique structural features, such as extended or kinked conformations and non-canonical disulfide bonds. The study demonstrates that filtered synthetic data can notably reduce structural violations and improve RMSD metrics.
3. The authors benchmark their Nanobody model against AlphaFold3 and NanobodyBuilder2, showing competitive performance with much faster inference times. The model achieves lower mean H-CDR3 RMSD while maintaining physical plausibility of predicted structures.
4. The study introduces a practical framework for optimizing VHH structure prediction, emphasizing the importance of stringent filtering criteria for synthetic data. This approach could facilitate large-scale screening of nanobody sequences in de novo design campaigns.
5. The results suggest promising directions for synthetic data generation in nanobody structure modeling. Combining this approach with optimized model architectures may further enhance prediction capabilities.
📜Paper: biorxiv.org/content/10.64898…
#Nanobody #StructurePrediction #SelfDistillation #SyntheticData #Bioinformatics #MachineLearning
2
18
1,447

20 Oct 2025
Major modes of behavior within low-pLDDT regions were identified through a survey of human proteome predictions provided by the AlphaFold Protein Structure Database @ActaCrystD @IUCr #AlphaFold2 #StructurePrediction #ConditionalFolding doi.org/10.1107/S20597983250…

2
4
19
2,267

📢 Save the Date!
The #ML4NGPevents calendar is out!
Join us as we explore how #MachineLearning is transforming research on non-globular #proteins, from #structureprediction to function.
📅 Mark your calendars — more info soon!
#ML4NGP #AI #Bioinformatics #ProteinResearch

1
3
87
23 Sep 2025
Pocket Restraints Guided by B-Cell Epitope Prediction Improves Chai-1 Antibody-Antigen Structure Modeling
1. A new study presents BepiPocket and DiscoPocket, two innovative methods that integrate B-cell epitope prediction tools to guide antibody-epitope restraints during Chai-1 structure prediction, significantly improving the accuracy and diversity of predicted antibody-antigen (AbAg) complexes.
2. The study demonstrates that using sequence-based predictor BepiPred-3.0 (BepiPocket) and structure-based predictor DiscoTope-3.0 (DiscoPocket) enhances AbAg structure prediction compared to standard Chai-1 modeling with random seed variation.
3. A key driver in the performance gains of BepiPocket and DiscoPocket is the antigen modeling accuracy. The study shows that better antigen modeling leads to more accurate and diverse AbAg complex predictions.
4. The research highlights the importance of reducing epitope redundancy in AbAg predictions. BepiPocket and DiscoPocket encourage Chai-1 to explore more diverse antibody binding sites, increasing the likelihood of identifying the correct binding site.
5. The study also investigates the impact of multiple sequence alignment (MSA) on AbAg structure prediction. Adding MSA input to Chai-1 substantially improves antigen modeling accuracy, which in turn enhances overall AbAg complex predictions.
6. The effectiveness of BepiPocket and DiscoPocket is robust even on independent test data released after the training cutoffs of the epitope prediction tools, demonstrating their potential for improving AbAg predictions in real-world applications.
7. The study concludes that BepiPocket and DiscoPocket provide a strong baseline for more accurate in silico prediction of antibody targets, with major applications in diagnostics and therapeutic antibody development.
📜Paper: biorxiv.org/content/10.1101/…
#AntibodyAntigen #StructurePrediction #BCellEpitope #ComputationalBiology #Bioinformatics

7
46
3,416
23 Sep 2025
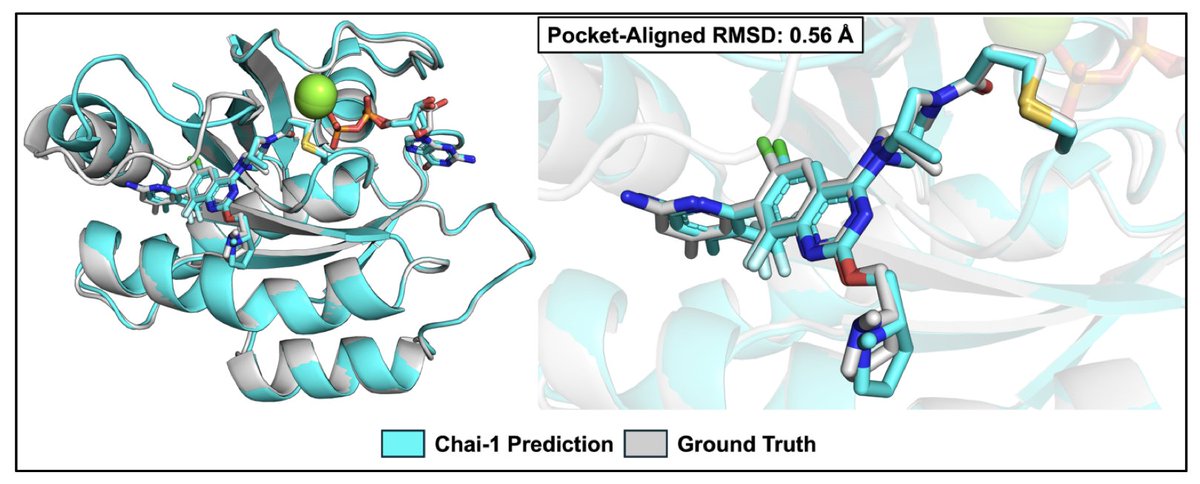
Single-sequence deep learning delivers crystal-quality models of covalent K-Ras G12 hotspot complexes
1. A novel study introduces Chai-1, a publicly available structure prediction tool that accurately predicts covalent K-Ras(G12C) complexes without using multiple sequence alignment (MSA). This tool achieves pocket-aligned RMSDs of less than 2 Å for a diverse range of K-Ras(G12C) inhibitors, demonstrating its high accuracy and potential for accelerating covalent drug discovery.
2. Chai-1 not only reproduces the binding poses of conventional acrylamide-based covalent K-Ras(G12C) inhibitors but also successfully models covalent K-Ras(G12D) and K-Ras(G12S) inhibitors using a covalent-bond restraint. This unique feature addresses the challenge of predicting interactions with less nucleophilic targets like aspartate and serine, expanding the scope of covalent drug design beyond cysteine.
3. The study highlights Chai-1's computational efficiency, offering approximately 40-fold higher throughput than AlphaFold3 while maintaining comparable pose accuracy. This efficiency makes Chai-1 a practical and scalable solution for large-scale binding pose prediction campaigns, significantly reducing the time and resources required for covalent drug development.
4. The research validates Chai-1's ability to predict covalent inhibitor poses for K-Ras(G12C), K-Ras(G12D), and K-Ras(G12S) at near-crystal resolution. However, it also identifies limitations, such as the model's bias towards abundant structures in the PDB and challenges in capturing fine-grained chemical details like bond angles and stereochemistry. These insights provide a clear direction for future improvements and applications.
5. The study's findings establish Chai-1 as an accessible and computationally efficient tool for covalent protein-ligand co-complex structure prediction. Its open-source nature and commercial usability under an Apache 2.0 license make it a valuable resource for the scientific community, enabling rapid and accurate structural insights for challenging covalent drug targets.
📜Paper: biorxiv.org/content/10.1101/…
#DeepLearning #CovalentInhibitors #KRas #StructurePrediction #Chai1 #DrugDiscovery
8
57
3,481
13 Sep 2025
Christopher J. Williams et al.: Categorizing prediction modes within low-pLDDT regions of AlphaFold2 structures: near-predictive, pseudostructure and barbed wire #AlphaFold2 #StructurePrediction #ConditionalFolding ... #IUCr journals.iucr.org/paper?S205…
2
6
520
28 Aug 2025
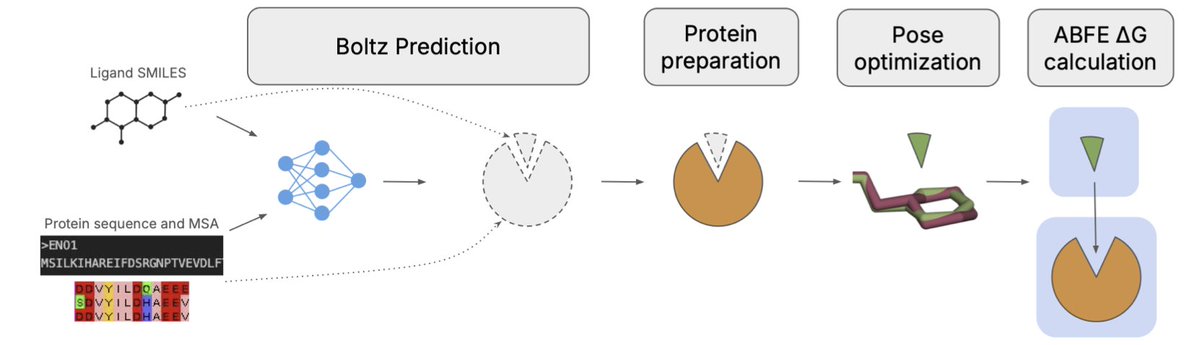
Boltz-ABFE: Free Energy Perturbation without Crystal Structures
1. A groundbreaking study introduces Boltz-ABFE, a novel pipeline that combines Boltz-2 structure prediction models with absolute free energy perturbation (ABFE) protocols to estimate small molecule binding affinity without relying on experimental crystal structures. This innovation significantly expands the applicability of FEP in early-stage drug discovery, where suitable crystal structures are often unavailable.
2. The study investigates the quality of protein-ligand complex structures predicted by Boltz-2 and proposes automated approaches to improve these structures for use in molecular dynamics simulations. The researchers demonstrate the effectiveness of Boltz-ABFE on four protein targets from the FEP benchmark set, achieving satisfactory results with mean unsigned errors (MUE) less than 1 kcal/mol on average.
3. A key innovation is the use of a coupled co-folding and re-docking approach to correct chemical inaccuracies in the predicted ligand structures. The researchers found that while Boltz-2 significantly reduces errors compared to Boltz-1, issues with stereochemistry still persist. They address this by re-docking the ligand into the predicted receptor using traditional docking software, which improves the accuracy of the ligand pose.
4. The study also explores the use of classical scoring functions for target deconvolution, finding that while these methods can be effective for targets with low sequence similarity, they are insufficient for more challenging cases with similar structures and ligand affinities. This highlights the importance of using Boltz-ABFE for accurate binding affinity predictions in complex scenarios.
5. The researchers emphasize the importance of careful sequence and biological assembly tailoring when generating co-folded structures for MD simulations. They demonstrate that truncating low-confidence regions and including binding partners can significantly improve model confidence scores and structural validity, leading to more accurate ABFE predictions.
6. The Boltz-ABFE pipeline is shown to be competitive with the Boltz-2 Affinity module, which is trained on experimental binding affinity data. However, the authors argue that Boltz-ABFE is more robust across different targets due to its physical foundation and bottom-up parametrization. Future improvements could include incorporating co-factors and optimizing the ABFE protocol to reduce the offset effect in early-stage drug discovery applications.
📜Paper: arxiv.org/abs/2508.19385v1
#BoltzABFE #FreeEnergyPerturbation #DrugDiscovery #MolecularDynamics #StructurePrediction
1
18
70
5,244
15 Aug 2025
Accelerating Biomolecular Modeling with AtomWorks and RF3
🚀 New preprint from David Baker!🚀
1. A new framework called AtomWorks has been introduced to revolutionize biomolecular modeling. AtomWorks provides a unified and modular platform for developing state-of-the-art biomolecular models, including structure prediction, protein design, and sequence design. It streamlines the process of data preparation and model training, making it easier for researchers to prototype and test new ideas.
2. The AtomWorks framework emphasizes high-quality data handling. It standardizes inputs from diverse sources, such as the Protein Data Bank (PDB), and resolves common issues like incorrect bond orders, charges, and missing coordinates. This results in higher-quality derived features and improved model performance. For example, AtomWorks-generated reference conformers have lower energies compared to those from other open-source models.
3. AtomWorks enables rapid prototyping by breaking down data processing and featurization into modular components. This modular design allows researchers to reuse core building blocks across different networks and easily add new features. It also simplifies the integration of various datasets, facilitating the training of models like RF3 on a diverse set of biomolecular structures.
4. The framework supports scalable training of biomolecular models. AtomWorks shares most of its code across different networks, allowing researchers to repurpose existing components and improve common operations. This efficiency is demonstrated by the ability to process large batches of data quickly, such as processing a 10,000-token batch through the LigandMPNN pipeline in the time it takes for a single forward/backward pass.
5. AtomWorks is accompanied by industry-grade testing and comprehensive documentation. This ensures that the framework is reliable and easy to use, even for researchers without extensive software development experience. The documentation includes worked examples illustrating how to develop pipelines for various biomolecular modeling tasks.
6. Using AtomWorks, the authors trained RosettaFold-3 (RF3), an all-atom biomolecular structure prediction network. RF3 incorporates novel features such as implicit chirality representations and atom-level geometric conditioning, which improve its performance on tasks like predicting chiral ligands and fixed-backbone conformations.
7. RF3 simplifies dataset integration by supporting direct loading from raw crystallographic information files (CIF). The authors introduced new distillation datasets, including a nucleic acid complex distillation set and an RNA distillation set, to enhance the model's training. Additionally, RF3 includes a disordered distillation set to address issues with hallucinated secondary structures.
8. RF3 accurately adheres to specified stereochemistry out-of-the-box, without requiring inference-time guidance. It represents stereochemistry by the sign of the angles formed by atoms surrounding each chiral center and uses data augmentation techniques to improve chirality handling. As a result, RF3 predicts the correct chirality for 88% of ligand chiral centers in the test set, compared to 84% for AlphaFold3 and 76% for Boltz-2.
9. RF3 enables flexible user control through arbitrary atom-level conditioning. Users can specify distances between atoms to incorporate experimentally derived constraints, perform protein-ligand docking, or fold proteins around specific ligand conformers. This feature significantly improves the accuracy of protein-ligand interface predictions.
10. RF3 narrows the performance gap between existing open-source structure prediction models and AlphaFold3. It demonstrates competitive performance on various tasks, such as predicting protein-protein interfaces, protein-ligand interactions, and mixed L/D peptides. When trained on data up to January 2024, RF3 shows further improvements in performance.
11. The authors also trained ProteinMPNN and LigandMPNN using AtomWorks, demonstrating comparable performance to the original models. This highlights the versatility of the AtomWorks framework in supporting different biomolecular modeling tasks.
12. The AtomWorks framework and RF3 model are released with curated training data, code, and model weights, making them accessible for further research and development in the field of biomolecular modeling.
💻Code: github.com/RosettaCommons/at…
📜Paper: biorxiv.org/content/10.1101/…
#BiomolecularModeling #AtomWorks #RF3 #StructurePrediction #ProteinDesign #OpenSource #MachineLearning #DeepLearning #ComputationalBiology

2
45
146
24,311
21 Jul 2025
Fold first, ask later: structure-informed function annotation of Pseudomonas phage proteins
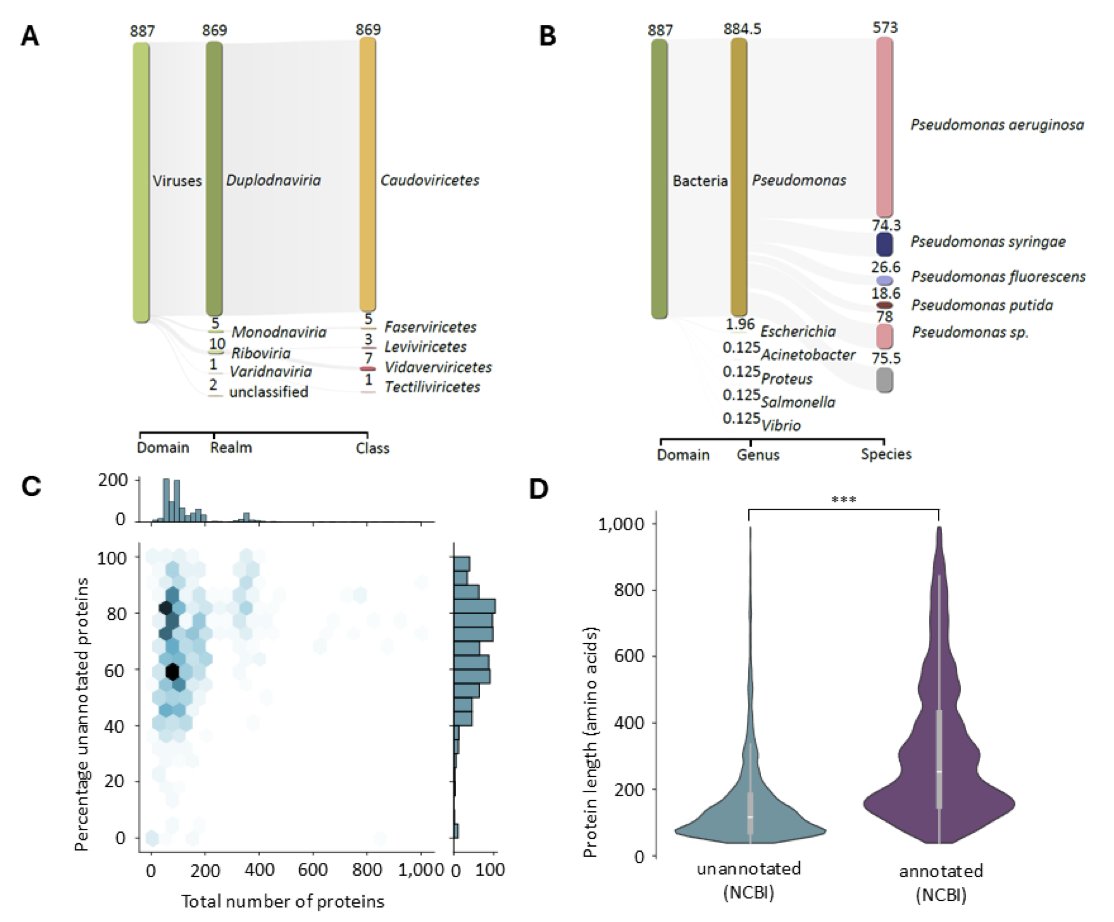
1. This study explores the “viral dark matter” of Pseudomonas infecting phages, focusing on annotating hypothetical proteins using structure-informed approaches. The authors curate a dataset of over 10,000 proteins and predict their structures with ColabFold, assessing structural similarity via FoldSeek against multiple databases.
2. The study finds that up to 43% of truly unannotated proteins can be functionally annotated by combining structure-informed approaches with UniProt-derived annotations. This highlights the power of leveraging protein structure to overcome the limitations of sequence-based annotation methods.
3. The authors demonstrate the complementarity of different databases (PDB, AlphaFold, and Phold) in annotating phage proteins. The PDB provides experimental evidence, while AlphaFold and Phold offer computational predictions, each contributing unique insights.
4. A three-tiered annotation classification scheme is introduced to improve annotation quality by distinguishing between high-information and low-information annotations. This method reduces the number of uninformative annotations and enhances the overall reliability of the annotations.
5. The study emphasizes the importance of annotation quality filtering and provides a valuable resource of predicted structures and annotations for Pseudomonas phage proteins. This work paves the way for deeper exploration of phage biology and its applications in biotechnology and medicine.
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/hannelorelongin/F…
#PhageProteins #ProteinAnnotation #StructurePrediction #Bioinformatics #ViralDarkMatter
3
796
5 Jul 2025
STRAND: Structure Refinement of RNA-Protein Complexes via Diffusion
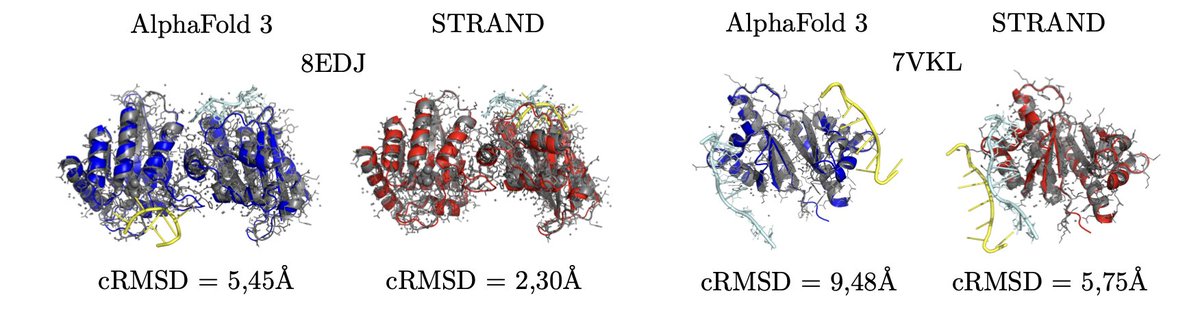
1.STRAND introduces a new diffusion-based method for refining RNA-protein complex structures, significantly improving initial predictions from AlphaFold 3 and ProRNA3D-single.
2.Unlike previous approaches focused on proteins, STRAND extends DiffDock-PP to handle RNA-protein interactions by incorporating RNA-specific features using embeddings from the RNA-FM foundation model.
3.STRAND improves the backbone positioning of predicted complexes by applying learned transformations—translation, rotation, and torsion—to the protein while treating the RNA as a fixed receptor.
4.Experimental results show that simple translation and rotation refinements alone often outperform or match the original predictions, indicating frequent global misplacements in initial models.
5.The best performance is achieved by STRANDtr rot (translation rotation), which lowers the mean cRMSD for AlphaFold 3 predictions from 3.09Å to 2.97Å on X-ray validated structures and improves predictions in 63% of non-X-ray cases.
6.The torsion-only model shows limited benefit, suggesting that global repositioning is more critical than internal flexibility for improving RNA-protein docking predictions.
7.STRAND consistently improves ProRNA3D-single outputs as well, reducing mean cRMSD from 12.02Å to 11.19Å and showing refinement in over 80% of tested cases.
8.A trained confidence model for sample selection allows STRAND to pick the best refined structures from multiple candidates, yielding further gains especially on lower-quality non-X-ray datasets.
9.Because STRAND acts as a refinement step, users can fall back on original structures when refinement is unhelpful, making it a robust plug-in module for existing prediction pipelines.
10.The model currently refines backbone structures only and is limited by sequence length restrictions from foundation models, but future work aims to expand to full-atom refinement and improve torsion modeling.
📜Paper: biorxiv.org/content/10.1101/…
#RNAProtein #StructurePrediction #DiffusionModel #AlphaFold3 #ComputationalBiology #STRAND
4
14
956