
New on bioRxiv | Generalisable tissue-wide molecular reconstruction from histology
A new AI method that reconstructs full tissue-level molecular profiles directly from routine histology images - bridging spatial transcriptomics & standard pathology at scale.
doi.org/10.64898/2026.06.09.…
#AIxBiology #SpatialTranscriptomics #Pathology #ComputationalBiology
6

Come hear Ben Hogan at #Angioscience26!
Ben uses high-resolution live imaging, forward genetic zebrafish screens 🐟 and single-cell transcriptomics to study lymphangiogenesis, the blood-brain barrier formation and pericyte maturation. 🔬
Deadline June 15th!

21

A new multiomics study finds that long COVID patients, months after hospital discharge, still carry blood signatures of vascular inflammation, platelet overactivation, and complement system dysfunction — the same pathways that made acute COVID-19 dangerous in the first place.
Severe COVID-19 triggers what researchers call immunothrombosis: a self-reinforcing loop in which complement activation primes neutrophils, neutrophils release DNA nets that trap and activate platelets, and platelets amplify coagulation and further complement signaling. In hospitalized patients, complement initiators C1QA, C1QB, and C1QC were among the most significantly upregulated genes, alongside platelet surface receptors indicating high aggregation potential. Simultaneously, infection-driven oxygen deprivation forced cells into Warburg-like metabolism, suppressing mitochondrial oxidative function and switching to glycolysis — with elevated acylcarnitines in urine confirming the shutdown of normal fat metabolism.
Three months out, long COVID patients look different:
A Latvian Biomedical Research and Study Centre study tracked 81 hospitalized patients across acute illness, one month, and three months using blood transcriptomics, urine metabolomics, and kidney injury markers. Most immune and metabolic pathways partially normalize across the cohort. But among the 46 patients who developed long COVID — older on average, higher BMI, longer hospitalization — a distinct transcriptional signature persisted at three months: sustained endothelial activation, residual platelet reactivity, complement dysregulation, and low-grade vascular inflammation not present in the 35 who recovered.
Kidneys and metabolism:
Urine profiling showed gradual but incomplete renal recovery. Acute kidney injury affected a significant share of the hospitalized cohort, consistent with prior findings of tubular injury and thrombotic microangiopathy in severe COVID-19. Mitochondrial repair signals — mitophagy regulators, fusion genes, heme biosynthesis pathways — showed a rebound trajectory through recovery, suggesting the body is still rebuilding oxidative capacity three months after discharge.
cell.com/iscience/fulltext/S…
10

📈 #TrendingWithImpact:
This #research paper was #published in Volume 18, titled "Single-cell transcriptomics reveal intrinsic and systemic T cell aging in COVID-19 and HIV."
#PressRelease ⬇️
aging-us.net/2026/02/19/new-…
#aging #HIV #COVID #Tcells #oa #journal #publishing #press
25
闫明 retweeted

Jun 12
Kingdom-wide comparative transcriptomics reveals deeply conserved and predictable stress response programs across Viridiplantae
biorxiv.org/content/10.64898…
♻️


1
8
31
1,682

1/5
A heat map is simply a table of numbers displayed as colors.
Each square represents a measurement.
In genomics and transcriptomics, those measurements are often gene expression levels.
Instead of reading thousands of numbers, you can instantly spot patterns using color.

1
19

What We Have Observed Regarding Spike-Associated Biological Damage
While research continues to evolve, a growing body of published literature, ongoing molecular investigations, and observations from active patient datasets have identified recurring patterns of biological disruption associated with persistent spike protein exposure and expression.
The evidence increasingly suggests that spike-associated injury is not confined to a single organ system. Rather, it appears capable of affecting multiple interconnected biological networks that govern resilience, repair, immune surveillance, metabolism, and cellular function.
In both published findings and active patient data, we have observed recurring patterns that include:
•Disruption of normal brain function, neuroinflammatory pathways, and neurovascular integrity.
•Injury to the heart, vascular endothelium, and microcirculation.
•Lung damage, inflammatory remodeling, fibrosis-related pathways, and impaired respiratory resilience.
•Kidney stress and disruption of normal tissue repair mechanisms.
•Alterations of the microbiome and weakening of protective microbial ecosystems that support immune defense.
•Disease-promoting shifts across multiple biological systems that reduce the body’s ability to maintain resilience and homeostasis.
•Mitochondrial dysfunction resulting in diminished cellular energy production, increased oxidative stress, and impaired recovery capacity.
•Dysregulation of immune surveillance, reducing the body’s ability to appropriately recognize, respond to, and eliminate biological threats.
•Accelerated biological aging characterized by persistent inflammatory and metabolic stress.
•Molecular conditions associated with more aggressive cancer behavior, clonal adaptation, progression, and loss of normal cellular regulatory control.
Taken together, these observations suggest that spike-associated biological injury should not be viewed merely as an isolated event or transient exposure. Rather, it appears capable of functioning as a systems-level biological disruptor that can influence numerous pathways involved in human health, resilience, recovery, and longevity.
At Neo7 Bioscience, our focus has been to move beyond symptom management and toward molecular interrogation of these disruptions. Through advanced transcriptomics, proteomics, molecular surveillance, and personalized peptide engineering, we have worked with individuals seeking to restore resilience pathways, improve biological function, and address the complex downstream consequences associated with spike-related molecular dysregulation.
The future of medicine is not simply diagnosing damage after it occurs—it is identifying molecular instability early and engineering precision interventions that restore biological resilience before dysfunction becomes disease.
@P_McCulloughMD @NicHulscher @McCulloughFund
Further Information:
neo7bioscience.com

What is Spike?
Sometimes the most important questions in medicine are the ones we’re still afraid to ask.
Science advances when curiosity is allowed to go further than consensus.
The spike protein has become one of the most discussed biological structures of our time, yet many fundamental questions remain regarding its interaction with inflammation, vascular function, immune regulation, cellular signaling, and long-term biological resilience.
As physicians, scientists, and innovators, our responsibility is not to defend narratives.
Our responsibility is to pursue understanding.
The future of healthcare will belong to those willing to investigate complex biological systems with intellectual honesty, scientific rigor, and an unwavering commitment to discovery.
Progress begins with questions.
And better questions lead to better medicine.
— Dr. John Catanzaro
7
16
754

🗣️ Excellent talk by Beatriz Martin at #ESHG2026!
🧬 “Multi-layer integration of long-read genome sequencing, transcriptomics and methylation outlier detection in 20 unsolved rare disease trios”
👏 Great work by the Functiontal Genomics team!
#RareDiseases #Genetics


1
120

🔬 MetaboRamics: Raman imaging for live-cell spatial metabolomics, revealing dynamic metabolic rewiring in EMT
📊 Single-cell foundation models plateau at 1–10% of current datasets
🧭 CytoSignal: Infers ligand-receptor signaling at cellular resolution from spatial transcriptomics
18
🚨Lightning Talk alert at #ESHG!
👉Discover the power of genomics for new #RareDiseases diagnoses: “Multi-layer integration of long-read genome sequencing, transcriptomics and methylation outlier detection in 20 unsolved rare disease trios”
📌 Lightning Talks Stage 1
⏰ 13:10

1
67

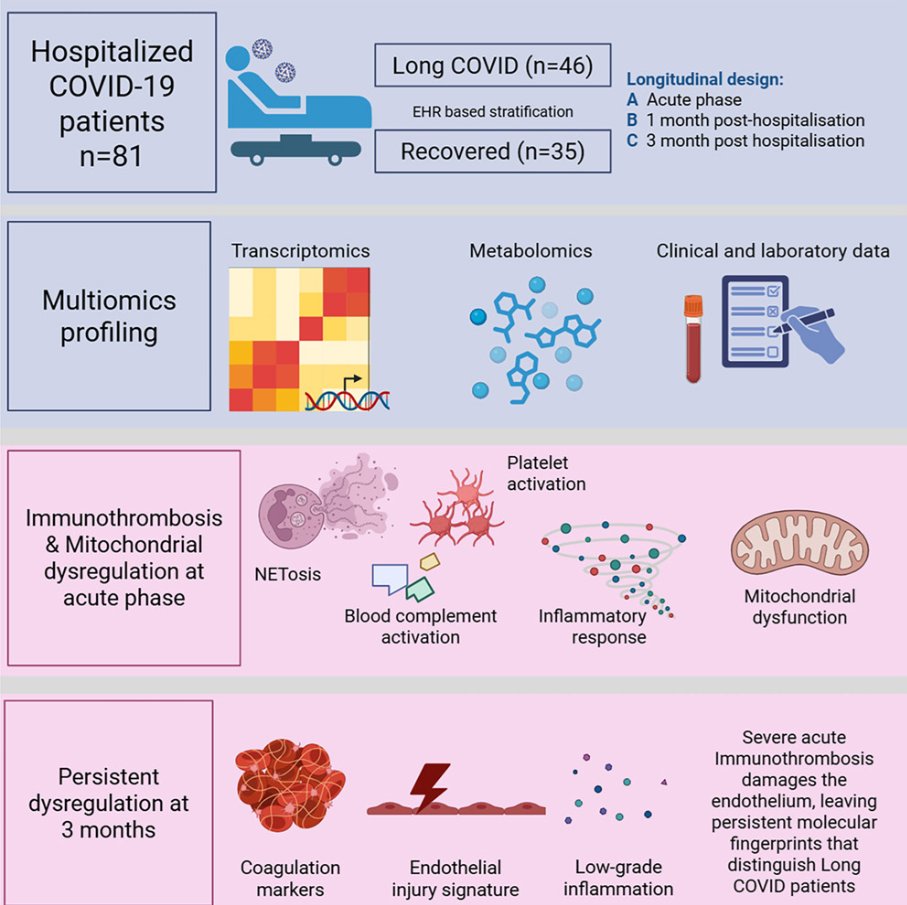
Immunothrombosis in hospitalized COVID-19 patients identified by multiomics profiling and linked to postacute complications
🚨INTERESTING New Latvian/Swedish multi-omics study shows immunothrombosis never fully switched off in longC0VID patients: 3 months after COVID hospitalization, your blood is still biologically “clot-ready.”
➡️What makes this study so important:
1. It reinforces that immunothrombosis (the interplay of complement, NETs, and platelets) is a central driver in severe acute COVID-19 and can persist in longC0VID,
2. It confirms persistent endothelial Dysfunction/Endotheliopathy in longC0VID patients, consistent with earlier studies on vascular damage and microclots,
3. It aligns with prior evidence of mitochondrial dysfunction during the acute phase, followed by partial repair mechanisms,
4. The persistent complement activation they observed fits with other recent multi-omics studies showing ongoing complement dysregulation in longC0VID.
➡️Study:
1. Prospective longitudinal multi-omics study of 81 hospitalized COVID-19 patients tracked whole-blood transcriptomics, urine metabolomics (46 analytes), and 13 kidney-injury biomarkers at acute admission, ~1 month, and ~3 months post-hospitalization,
2. Patients stratified by EHR into recovered (n=35) versus long COVID (n=46) groups based on a PASC diagnoses within 12 months,
3. None of the 81 hospitalized patients were vaccinated,
4. Acute phase dominated by interconnected immunothrombosis: strong upregulation of complement (C1QA/B/C), NETosis (PADI4, MPO), and platelet-activation genes (ITGA2B, ITGB3), plus mitochondrial dysfunction (HIF1A/EPAS1 up, OXPHOS down, Warburg-like glycolysis) and elevated renal injury markers (KIM-1 etc.),
5. Most immune, mitochondrial, and metabolomic changes largely normalized by 1–3 months, with rebound in mitophagy/heme genes (PINK1, OPA1, FECH) indicating repair,
6. At 3 months, longC0VID patients showed a distinct transcriptional signature of persistent endothelial activation (↑VWF, PROS1, ITGA2B/ITGB3), complement dysregulation (CFH), and low-grade vascular inflammation/platelet reactivity (CXCL5, ALOX12) that was absent in recovered individuals,
7. No significant late differences in urine metabolomics or kidney biomarkers between groups.
➡️They conclude with their Highlight-points:
• Severe COVID-19 induces immunothrombosis-associated molecular programs,
• Acute COVID-19 is associated with mitochondrial metabolic dysregulation,
• Urine profiling indicates gradual renal recovery after hospitalization,
• LongC0VID patients retain endothelial-associated activation signatures.
‼️So, even after apparent clinical recovery, immunothrombosis leaves a persistent molecular scar of endothelial activation and prothrombotic signalling in longC0VID patients at three months, revealing that the acute vascular battlefield never fully quiets in those who remain symptomatic.
→Three months post Covid-19, longC0VID patient’s blood is still biologically primed to clot!
#AvoidSars2 #AvoidReinfections
cell.com/iscience/fulltext/S…
4
30
77
2,437

With OmicsLogic, you can explore Genomics, Transcriptomics, and Metagenomics through our Multi-Omics Course Track: omicslogic.com/programs/trac…

12

13h
A fascinating new study reframes idiopathic inflammatory myopathies (IIMs): the key drivers of chronic muscle inflammation may not be immune cells alone, but tissue-resident fibro-adipogenic progenitors (FAPs).
Using single-nucleus RNA-seq, spatial transcriptomics, ATAC-seq, and primary human FAP cultures, researchers analyzed muscle biopsies from anti-synthetase syndrome (ASYS), inclusion body myositis (IBM), and immune-mediated necrotizing myopathy (IMNM). They found that FAPs adopt disease-specific inflammatory phenotypes that mirror the dominant immune environment.
Key findings:
🔹 IBM FAPs acquired T-cell–oriented programs, expressing mediators such as IL7 and CCL13.
🔹 IMNM FAPs preferentially engaged macrophage-associated inflammatory pathways.
🔹 ASYS FAPs displayed humoral immunity signatures and elevated IL6 expression.
🔹 Across all IIM subtypes, FAPs lost homeostatic markers (e.g., COL15A1) and shifted toward pro-inflammatory and pro-fibrotic states.
Trajectory analysis revealed two major FAP fates:
Homeostatic FAPs supporting muscle structure and regeneration.
Pro-inflammatory FAPs characterized by CXCL1, CCL2, IL18, LIF, COL1A1, COL1A2, LOX, and extracellular matrix remodeling programs.
Importantly, the proportion of pro-inflammatory FAPs increased with disease duration, suggesting progressive stromal reprogramming during chronic inflammation.
Mechanistically, the study identifies a dual-input signaling axis:
• Immune cells provide TGF-β signals.
• Damaged myofibers provide EGF signals.
Both converge on AP-1 transcription factor activity (JUN/FOS family), driving chromatin remodeling and establishment of a pathogenic FAP state. ATAC-seq demonstrated increased AP-1 accessibility after TGF-β EGF stimulation, while AP-1 inhibition reduced inflammatory cytokine induction.
Spatial transcriptomics further showed that FAPs form inflammatory niches by co-localizing with macrophages, muscle stem cells, and—in IBM—T cells, positioning them as organizers of local immune microenvironments rather than passive bystanders.
The broader implication is significant: chronic autoimmune muscle disease may involve a form of "stromal memory" or tissue priming, analogous to pathogenic fibroblasts in rheumatoid arthritis. If so, targeting FAP reprogramming, AP-1 signaling, BET proteins, or TGF-β pathways could complement conventional immunosuppression and potentially address treatment-refractory disease.
Reference
Nelke C et al. Inflammation reprograms fibro-adipogenic progenitors to sustain immunopathogenic niches in myositis. Cell Death & Disease (2026). DOI: 10.1038/s41419-026-08966-w.

2
92
Dr S S Hasan retweeted

Jun 5
Important paper in Gut! 📢 New study maps the evolution of pancreatic cysts to cancer
Cui et al. used spatial transcriptomics to map progression from IPMNs to pancreatic ductal adenocarcinoma—revealing it’s not linear, but follows distinct molecular paths.
Early-stage lesions showed active immune surveillance (T cells and NK cells), while progression was marked by a shift to an immunosuppressive environment with expansion of regulatory T cells and fibroblasts.
Intriguingly, histologically similar high-grade lesions with different malignant potential were found to be molecularly distinct.
Malignant trajectories were characterised by upregulation of invasion-associated genes (e.g. CLDN1) TGF-β / IL-17 signalling.
⚖️ Are we over-treating some IPMNs while missing aggressive disease in others?
This study highlights the limitations of current risk stratification and the promise of spatially resolved biomarkers.
Check the paper: doi: bit.ly/4nIDprm
Social Media summary by Dr John P Thomas, Chain-Florey Clinical Research Fellow, Imperial College London, London, UK

3
6
606
Dr S S Hasan retweeted
Jun 12
Important paper in Gut! 📢 New study maps the evolution of pancreatic cysts to cancer
Cui et al. used spatial transcriptomics to map progression from IPMNs to pancreatic ductal adenocarcinoma—revealing it’s not linear, but follows distinct molecular paths.
Early-stage lesions showed active immune surveillance (T cells and NK cells), while progression was marked by a shift to an immunosuppressive environment with expansion of regulatory T cells and fibroblasts.
Intriguingly, histologically similar high-grade lesions with different malignant potential were found to be molecularly distinct.
Malignant trajectories were characterised by upregulation of invasion-associated genes (e.g. CLDN1) TGF-β / IL-17 signalling.
⚖️ Are we over-treating some IPMNs while missing aggressive disease in others?
This study highlights the limitations of current risk stratification and the promise of spatially resolved biomarkers.
Check the paper: doi: bit.ly/4nIDprm
Social Media summary by Dr John P Thomas, Chain-Florey Clinical Research Fellow, Imperial College London, London, UK

4
7
644

Cho et al. integrated whole-section (WS) spatial transcriptomics across 12 cancer types to construct a pan-cancer tertiary lymphoid structure (TLS) atlas. TLSs spanned early, primary, and secondary maturation states with distinct spatial niches and immune organization. Tumor regions proximal to intratumoral TLSs showed enriched antigen-presentation and IFN-response programs, and reduced proliferative and EMT signatures. An AI framework trained on whole-slide H&E images classified TLS maturation and maturation-aware TLS composite scores, which stratified survival and treatment response, outperforming conventional TLS metrics. bit.ly/4dZAbwi @mdAndersonNews @IamLinghua

5
23
1,259
antisense. retweeted

10 Jun 2025
PhenoGraph: A Multi-Agent Framework for Phenotype-driven Discovery in Spatial Transcriptomics Data Augmented with Knowledge Graphs biorxiv.org/content/10.1101/…

1
25
85
5,379
antisense. retweeted
7 Jun 2025
CellNEST reveals cell–cell relay networks using attention mechanisms on spatial transcriptomics nature.com/articles/s41592-0…

1
5
16
1,359